
MDR-Praxis, Teil III: Analyse Ihrer aktuellen Technischen Dokumentation
09.07.2019
Was bisher geschah:
Im ersten Teil unserer Reihe zur Umstellung der Bestandsprodukte auf MDR-Niveau haben wir die Unterschiede zwischen den bekannten Anforderungen der MDD zu den teils neuen bzw. präzisierten Anforderungen der MDR unter die Lupe genommen: Was genau ist neu daran? Was ist anders?
Mit dem zweiten Teil zur MDR-Praxis haben wir die Basis für weitere Dokumentationsschritte geschaffen: Sie haben Ihr Medizinprodukt spezifiziert und definiert, jetzt kennen Sie auch Ihr Zubehör und die weiteren Produkte, mit denen Ihr Produkt gemeinsam seine Funktion erbringt. Aus regulatorischer Sicht hat Ihr Produkt nun eine Zweckbestimmung und Sie haben die Produktklasse gemäß Artikel 51 („Klassifizierung von Produkten“) bzw. Anhang VIII festgelegt.Heute wollen wir herausarbeiten, welche Grundlegenden Sicherheits- und Leistungsanforderungen für Ihr Produkt tatsächlich neu sind. Ausgangspunkt für diese Analyse ist der Nachweis Ihrer Produktkonformität mit der MDD, Anhang I.Dazu springen wir kurz zurück zum ersten Teil: Mit Umsetzung des Beitrags war nämlich klar, in welche Kategorien die Anforderungen aus Anhang I eingeteilt werden können:
Ihre Aufgabe ist es nun, die bestehende Technische Dokumentation, die Ihnen als Nachweis zur Konformität mit der MDD gedient hat, zu analysieren. Ziel unserer heutigen Etappe ist es, nach erfolgreichem Review einen Plan zur Umsetzung der zuvor definierten Maßnahmen erstellen zu können. Nächstes Mal werden wir darauf aufbauend den entsprechenden und dringend notwendigen Projektplan entwickeln:
Und damit zu unserem heutigen Thema:
Um herauszufinden, wie geeignet Ihre vorhandene Technische Dokumentation ist, fangen Sie mit den Grundlegenden Sicherheits- und Leistungsanforderungen an:
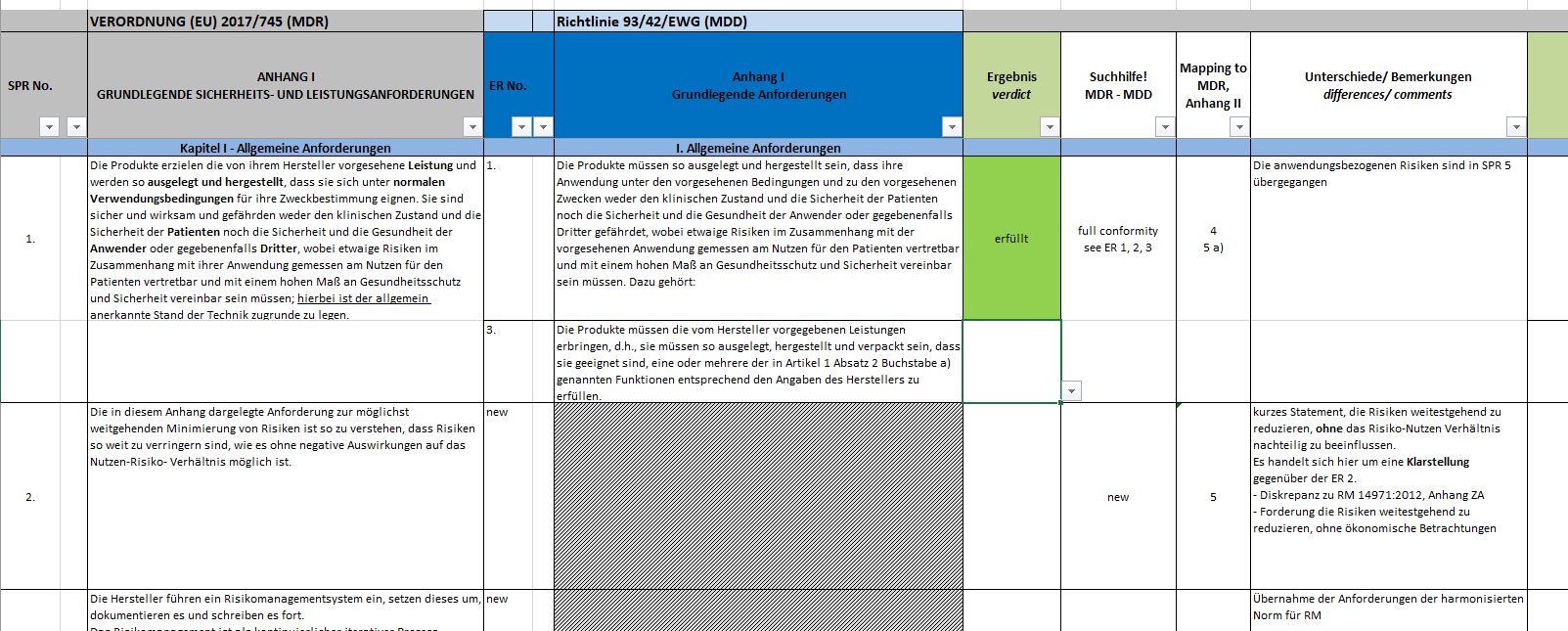
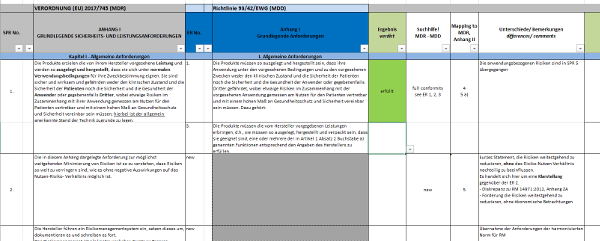 Klicken Sie hier für eine größere Ansicht.Ausgangspunkt ist eine Checkliste zum Nachweis der Erfüllung der Grundlegenden Sicherheits- und Leistungsanforderungen. Sofern diese noch nicht auf das Produkt angewendet wurde, prüfen sie jetzt, welche Anforderung anwendbar ist und welche nicht.TIPP: Die Anforderungen 1 bis 8 sind auf alle Produkte anwendbar. Wenn Sie von Anforderung 9 betroffen sind, rufen Sie gerne an, denn dann haben Sie vermutlich zum ersten Mal mit dem Regelwerk für Medizinprodukte zu tun. :-)Im nächsten Schritt ordnen Sie Ihre Nachweisdokumente den einzelnen Grundlegenden Sicherheits- und Leistungsanforderungen zu. Folgende Fragen sind jetzt zu beantworten:
Klicken Sie hier für eine größere Ansicht.Ausgangspunkt ist eine Checkliste zum Nachweis der Erfüllung der Grundlegenden Sicherheits- und Leistungsanforderungen. Sofern diese noch nicht auf das Produkt angewendet wurde, prüfen sie jetzt, welche Anforderung anwendbar ist und welche nicht.TIPP: Die Anforderungen 1 bis 8 sind auf alle Produkte anwendbar. Wenn Sie von Anforderung 9 betroffen sind, rufen Sie gerne an, denn dann haben Sie vermutlich zum ersten Mal mit dem Regelwerk für Medizinprodukte zu tun. :-)Im nächsten Schritt ordnen Sie Ihre Nachweisdokumente den einzelnen Grundlegenden Sicherheits- und Leistungsanforderungen zu. Folgende Fragen sind jetzt zu beantworten:
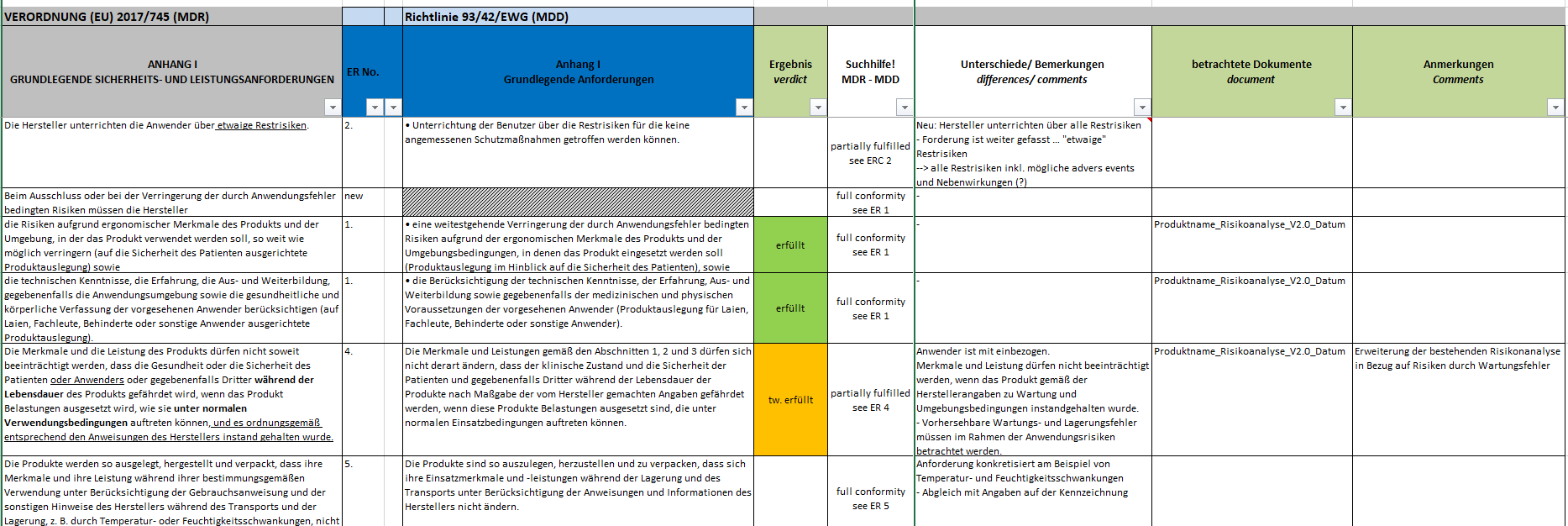
Sofern die Nachweise nicht ausreichen, sind die Lücken zu beschreiben und gegebenenfalls entsprechende Maßnahmen zu definieren.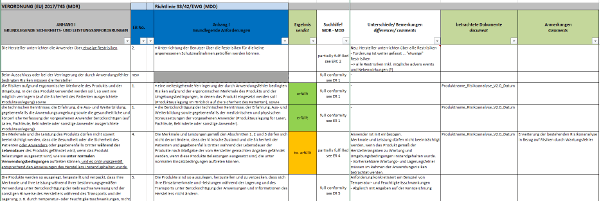 Klicken Sie hier für eine größere Ansicht.Kurz zusammengefasst gibt es bis hierher folgendes zu tun:
Klicken Sie hier für eine größere Ansicht.Kurz zusammengefasst gibt es bis hierher folgendes zu tun:
TIPP: Fehlen Ihnen Testberichte, sollten Sie diese zügig in Ihren eigenen Laboren oder mit externen Laboren planen. Beachten Sie dabei, dass Tests zur Biologischen Sicherheit, chemische Analysen, Sterilisationsvalidierungen und Co. meist mehrere Wochen Zeit für die Durchführung benötigen.
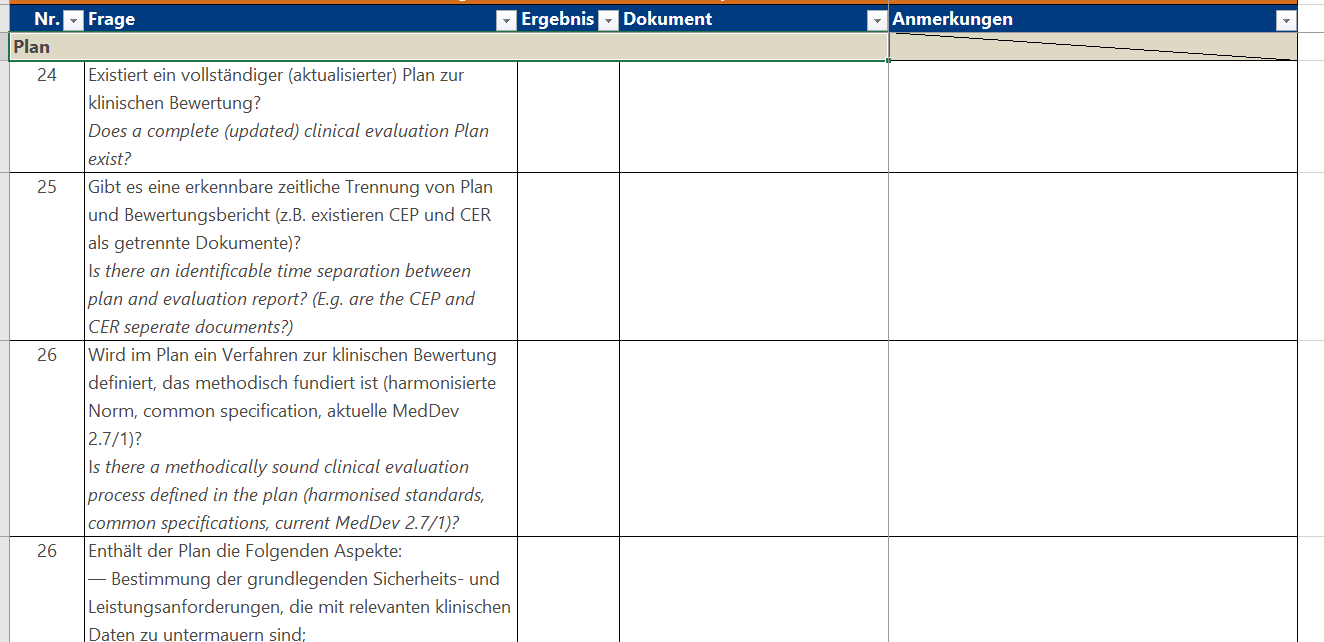
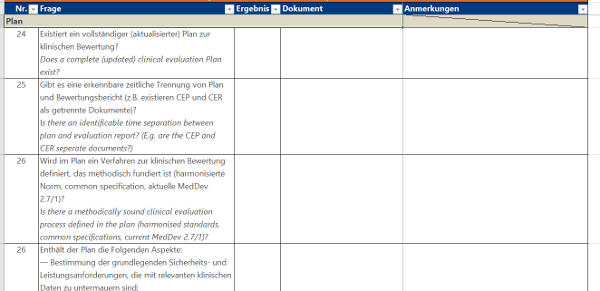 Klicken Sie hier für eine größere Ansicht.Einen Fragenkatalog in dieser Form können Sie ähnlich aufbauen wie Ihr Dokument zur Klinischen Bewertung mit folgenden Bausteinen:
Klicken Sie hier für eine größere Ansicht.Einen Fragenkatalog in dieser Form können Sie ähnlich aufbauen wie Ihr Dokument zur Klinischen Bewertung mit folgenden Bausteinen:
Ansonsten gehen Sie hier genauso vor wie bei der GAP-Analyse von Anhang I: Durchführung einer inhaltlichen Bewertung Ihrer Klinischen Bewertung in Bezug auf die Anforderungen der MDR, der Beschreibung der Feststellungen und der Definition von Maßnahmen.
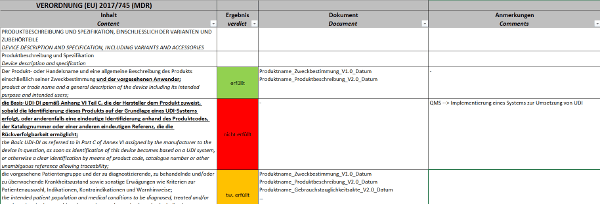 Klicken Sie hier für eine größere Ansicht.
Klicken Sie hier für eine größere Ansicht.
Wir können Ihnen nicht alles abnehmen, aber Ihr Team effizient unterstützen - in welchem Umfang, entscheiden Sie. Wir freuen uns auf Ihren Anruf!Nächstes Mal entwickeln wir dann die konkrete Strategie zur Überarbeitung der Technischen Dokumentation. Wir priorisieren die anstehenden Aufgaben und überlegen gemeinsam, womit Sie am besten anfangen sollten. Und ob Sie die Struktur Ihrer Technischen Dokumentation verändern müssen oder beibehalten können. Sind Sie wieder dabei?Viel Erfolg bei der Umsetzung und bis bald!
Ihre Sonja Bruhn
Sonja ist ein Metecon-Urgestein und seit Beginn im Thema Technische Dokumentation zuhause. Im Laufe der Jahre hat sie zahlreiche Kundenprojekte begleitet, so dass sie mittlerweile über einen reichen Erfahrungs- und Wissenschatz in allen Belangen des oft komplexen Themas Dokumentation und Marktzugang verfügt.Ihr Kontakt zu Sonja:
E-Mail: sonja.bruhn@metecon.de
T: +49 621 123469-16
Im ersten Teil unserer Reihe zur Umstellung der Bestandsprodukte auf MDR-Niveau haben wir die Unterschiede zwischen den bekannten Anforderungen der MDD zu den teils neuen bzw. präzisierten Anforderungen der MDR unter die Lupe genommen: Was genau ist neu daran? Was ist anders?
Mit dem zweiten Teil zur MDR-Praxis haben wir die Basis für weitere Dokumentationsschritte geschaffen: Sie haben Ihr Medizinprodukt spezifiziert und definiert, jetzt kennen Sie auch Ihr Zubehör und die weiteren Produkte, mit denen Ihr Produkt gemeinsam seine Funktion erbringt. Aus regulatorischer Sicht hat Ihr Produkt nun eine Zweckbestimmung und Sie haben die Produktklasse gemäß Artikel 51 („Klassifizierung von Produkten“) bzw. Anhang VIII festgelegt.Heute wollen wir herausarbeiten, welche Grundlegenden Sicherheits- und Leistungsanforderungen für Ihr Produkt tatsächlich neu sind. Ausgangspunkt für diese Analyse ist der Nachweis Ihrer Produktkonformität mit der MDD, Anhang I.Dazu springen wir kurz zurück zum ersten Teil: Mit Umsetzung des Beitrags war nämlich klar, in welche Kategorien die Anforderungen aus Anhang I eingeteilt werden können:
- Die Grundlegende Sicherheits- und Leistungsanforderung ist bekannt und auf eine Grundlegende Anforderung der MDD zurückzuführen.
- Die Grundlegende Sicherheits- und Leistungsanforderung ist teilweise bekannt und präzisiert lediglich eine bereits bekannte Anforderung der MDD.
- Die Grundlegende Sicherheits- und Leistungsanforderung ist neu und hat keine entsprechende Anforderung unter der MDD.
Ihre Aufgabe ist es nun, die bestehende Technische Dokumentation, die Ihnen als Nachweis zur Konformität mit der MDD gedient hat, zu analysieren. Ziel unserer heutigen Etappe ist es, nach erfolgreichem Review einen Plan zur Umsetzung der zuvor definierten Maßnahmen erstellen zu können. Nächstes Mal werden wir darauf aufbauend den entsprechenden und dringend notwendigen Projektplan entwickeln:
Maßnahmenplan + Verantwortlichkeiten + „realistische“ Zieltermine = Projektplan
TIPP: Je nach Produktumfang in Ihrem Unternehmen können Sie noch folgende strategische Überlegungen anstellen:- Habe ich ein Einzelprodukt?
- Habe ich Produktgruppen, die sich ähneln und für die ich einen „Piloten“ erstellen kann, den ich dann auf ähnliche Produkte übertragen kann? In diesem Fall ist das Ziel, mittels dieses Pilotprojekts eine Art Leitfaden zu generieren, mit dessen Hilfe die Kollegen dann weitere Produkte aus dem Portfolio analysieren können.
Und damit zu unserem heutigen Thema:
Ihre GAP-Analyse der Technischen Dokumentation
Um herauszufinden, wie geeignet Ihre vorhandene Technische Dokumentation ist, fangen Sie mit den Grundlegenden Sicherheits- und Leistungsanforderungen an:
a) Anhang I – Grundlegende Sicherheits- und Leistungsanforderungen
- Auswahl der anzuwendenden Grundlegenden Sicherheits- und Leistungsanforderungen
- Vergleich MDD – MDR und Erkennen der Unterschiede
- Bewertung der Nachweisführung zur Erfüllung der Anforderungen
Klicken Sie hier für eine größere Ansicht.Ausgangspunkt ist eine Checkliste zum Nachweis der Erfüllung der Grundlegenden Sicherheits- und Leistungsanforderungen. Sofern diese noch nicht auf das Produkt angewendet wurde, prüfen sie jetzt, welche Anforderung anwendbar ist und welche nicht.TIPP: Die Anforderungen 1 bis 8 sind auf alle Produkte anwendbar. Wenn Sie von Anforderung 9 betroffen sind, rufen Sie gerne an, denn dann haben Sie vermutlich zum ersten Mal mit dem Regelwerk für Medizinprodukte zu tun. :-)Im nächsten Schritt ordnen Sie Ihre Nachweisdokumente den einzelnen Grundlegenden Sicherheits- und Leistungsanforderungen zu. Folgende Fragen sind jetzt zu beantworten:- Haben Sie Nachweisdokumente (interne Testberichte, Testberichte aus externen Laboren, sonstige Referenzdokumente)?
- Reichen diese Dokumente aus, die Einhaltung der Anforderung umfassend zu belegen?
Sofern die Nachweise nicht ausreichen, sind die Lücken zu beschreiben und gegebenenfalls entsprechende Maßnahmen zu definieren.
Klicken Sie hier für eine größere Ansicht.Kurz zusammengefasst gibt es bis hierher folgendes zu tun:- die zutreffenden Grundlegenden Sicherheits- und Leistungsanforderungen auswählen,
- die vorhandenen Nachweisdokumente zuordnen,
- die vorhandenen Nachweisdokumente inhaltlich bewerten,
- die Feststellung/Lücke beschreiben,
- die Maßnahmen definieren.
TIPP: Fehlen Ihnen Testberichte, sollten Sie diese zügig in Ihren eigenen Laboren oder mit externen Laboren planen. Beachten Sie dabei, dass Tests zur Biologischen Sicherheit, chemische Analysen, Sterilisationsvalidierungen und Co. meist mehrere Wochen Zeit für die Durchführung benötigen.
b) Artikel 61 und Anhang XIV – Klinische Bewertung
Für die Klinische Bewertung existiert zwar keine fertige Anforderungsliste wie in Anhang I, aber der Artikel 61 in Kombination mit Anhang XIV definieren die Anforderungen an die Klinische Bewertung. Auch hier gilt wieder: Haben Sie nur einige wenige Produkte, können Sie Ihre vorhandenen Klinischen Bewertungen anhand der MDR reviewen. Ansonsten empfehle ich Ihnen die Anfertigung und Nutzung einer Checkliste, die Sie auch für weitere Produkte verwenden können. Für spätere Reviews von neu erstellten Klinischen Bewertungen kann eine solche Checkliste ebenfalls hilfreich sein.Klicken Sie hier für eine größere Ansicht.Einen Fragenkatalog in dieser Form können Sie ähnlich aufbauen wie Ihr Dokument zur Klinischen Bewertung mit folgenden Bausteinen:- Plan,
- Auswertung,
- besondere Anforderungen an Klasse III-Produkte und Implantate,
- Vergleichsprodukt,
- Fazit.
Ansonsten gehen Sie hier genauso vor wie bei der GAP-Analyse von Anhang I: Durchführung einer inhaltlichen Bewertung Ihrer Klinischen Bewertung in Bezug auf die Anforderungen der MDR, der Beschreibung der Feststellungen und der Definition von Maßnahmen.
c) Anhang II – Bewertung der Technischen Dokumentation auf Vollständigkeit
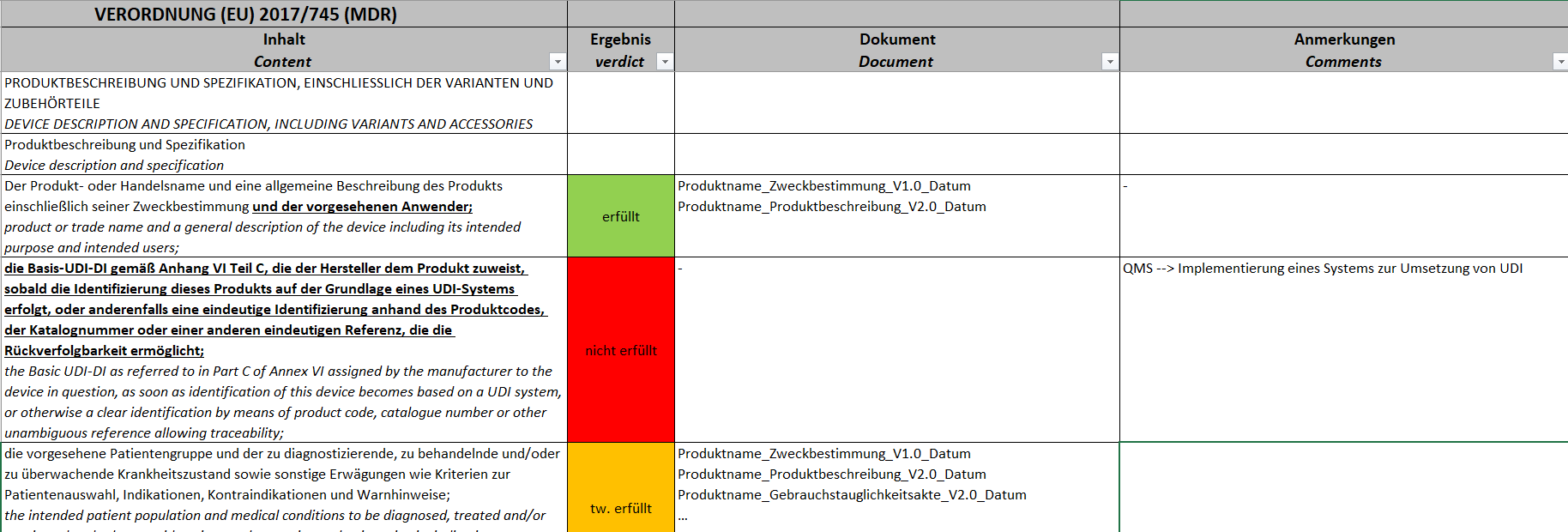
Anhang II spezifiziert sehr schön, welche Inhalte Ihre Technische Dokumentation enthalten muss. In welchen Dokumenten Sie diese Inhalte berücksichtigt haben, wird Ihnen nicht vom Gesetzgeber vorgegeben, hier sind Sie frei im Aufbau Ihrer Technischen Dokumentation. Prüfen Sie also, ob die geforderten Inhalte Bestandteil Ihrer Technischen Dokumentation sind. Teilweise werden Sie eigene Dokumente haben, teilweise werden die einzelnen Punkte Bausteine in Dokumenten sein.Es reicht nicht aus, nur Anhang I zu prüfen. Anhang II definiert Inhalte, die Sie nicht in Anhang I referenzieren werden, die aber zukünftig Bestandteil Ihrer Technischen Dokumentation sind.Ein Beispiel zur Umsetzung:Klicken Sie hier für eine größere Ansicht.d) Anhang III – Bewertung der Technischen Dokumentation (PMS) auf Vollständigkeit
Zuletzt schauen Sie sich Ihre Dokumentation zur Überwachung nach dem Inverkehrbringen an. Wie auch bei den anderen Anhängen bietet sich eine Checkliste an, nach der Sie Ihren PMS-Plan und den PMS-Bericht auf Vollständigkeit prüfen können. Unserer Erfahrung nach liegt dieser Teil der Dokumentation oft nicht ausreichend vor. Ein Grund dafür mag sein, dass die vorhandene Dokumentation in den seltensten Fällen den Anforderungen der MDD und Guidance Dokumenten genügt und – weil der Aufwand dafür hoch oder die Expertise noch nicht vorhanden ist? – nur lückenhaft umgesetzt ist. Wie sieht es bei Ihnen aus? :-)Herzlichen Glückwunsch, wieder ein Etappenziel erreicht: Nach erfolgreich durchgeführter GAP-Analyse der einzelnen Teile a) bis d) Ihrer Technischen Dokumentation kennen Sie jetzt die Lücken und sind in der Lage, konkrete Maßnahmen zu definieren.Zuletzt noch einige entscheidende Fragen in diesem Zusammenhang, auf die Sie zügig Antworten finden sollten:- Wie lange benötigen Sie für die Überarbeitung einer Technischen Dokumentation?
- Wie viele Technische Dokumentationen stehen bei Ihnen zur Überarbeitung an? Und wie lange benötigen Sie dann für ALLE zu überarbeitenden Technischen Dokumentationen?
- Haben Sie einen Transferplan, welche Produkte ggf. noch eine Zertifikatsverlängerung unter der MDD bekommen? Eventuell haben Sie diesen mit Ihrer Benannten Stelle festgelegt, dann können Sie jetzt stufenweise Ihre Bestandsprodukte zur MDR transferieren.
- Wenn Sie das noch nicht getan haben, aber über diese Möglichkeit nachdenken, sollten Sie sich sofort mit Ihrer Benannten Stelle in Verbindung setzen, um in Erfahrung zu bringen, wie lange diese noch Prüfungen unter MDD durchführt, und entsprechende Zeitfenster reservieren.
Wir können Ihnen nicht alles abnehmen, aber Ihr Team effizient unterstützen - in welchem Umfang, entscheiden Sie. Wir freuen uns auf Ihren Anruf!Nächstes Mal entwickeln wir dann die konkrete Strategie zur Überarbeitung der Technischen Dokumentation. Wir priorisieren die anstehenden Aufgaben und überlegen gemeinsam, womit Sie am besten anfangen sollten. Und ob Sie die Struktur Ihrer Technischen Dokumentation verändern müssen oder beibehalten können. Sind Sie wieder dabei?Viel Erfolg bei der Umsetzung und bis bald!
Ihre Sonja Bruhn
Sonja ist ein Metecon-Urgestein und seit Beginn im Thema Technische Dokumentation zuhause. Im Laufe der Jahre hat sie zahlreiche Kundenprojekte begleitet, so dass sie mittlerweile über einen reichen Erfahrungs- und Wissenschatz in allen Belangen des oft komplexen Themas Dokumentation und Marktzugang verfügt.Ihr Kontakt zu Sonja:
E-Mail: sonja.bruhn@metecon.de
T: +49 621 123469-16

 der MDCG")

{kind=link}
{kind=link}
{kind=link}
{kind=link}