
Mastering Your Product Lifecycle Reporting - Getting it right under the MDR
18/06/2024
Do you have any questions about the article or would you like to find out more about our services? We look forward to hearing from you!Make a non-binding enquiry now
With the Medical Device Regulation (Regulation (EU) 2017/745 (MDR)) and the accompanying guidelines of the Medical Device Coordination Group (MDCG), numerous obligations arise throughout the entire product lifecycle for you as a medical device manufacturer. These include the regular preparation of reports and, if necessary, updating of plans for post-market surveillance (PMS) and post-market clinical follow-up (PMCF), as well as for risk management and clinical evaluation. Some update timelines are mandatory, while others must be sensibly integrated into this cycle. To meet all the requirements within this "Product Lifecycle Reporting" and provide all documents on time, the individual phases must be well coordinated.
Risk Management accompanies the entire product lifecycle from the product idea to the end of life of the last item on the market. It must identify all risks, establish acceptance criteria, and assess the residual risk remaining after risk control measures have been implemented. To do this, risk management needs validation data such as that determined by the clinical evaluation.The Post-Market Surveillance (PMS) system accompanies the product lifecycle from market entry. Among other things, it must continuously ensure that new, changed or emerging risks are identified at all times, and - if necessary – that it is adapted accordingly and urgent measures are initiated immediately. The latter may include product changes and field measures.
From the first product idea and over the entire product development, the collection of clinical data must be planned. These data must then be generated and analyzed. Following the clinical evaluation report (CER), the PMCF (Post-Market Clinical Follow-up) plan is created. For CE marking, the PMS plan must also be created, and Class III products and implants additionally require an initial SSCP (Summary of Safety and Clinical Performance).For legacy devices, it is important to check that all documents are available before trying to achieve CE marking under the MDR. Not all reports were required under the Medical Device Directive (MDD - Directive 93/42/EEC on medical devices) nor during the transition phase, which is why they might not be available. This mainly concerns the SSCP – and often the clinical evaluation is too minimalistic. However, these reports must now exist and be compliant for certification under the MDR. The available data for legacy devices must be sufficient from the point of view of the MDR and in accordance with the Medical Device Coordination Group’s guidance MDCG 2020-6.As the requirements for a PMS system in accordance with the MDR, including vigilance, and its documentation already apply to legacy devices, these plans and reports should already be available for legacy devices when it is time for certification under the MDR.
After successful CE marking and market entry, the data collection period that you will need to follow is determined by the risk class of your device. You will have a data collection period of 1 year for Class IIb and III medical devices, 2 years for Class IIa devices, or until a report is needed (Class I). During this period, the planned PMS and PMCF activities are carried out, data are collected , and records created. Naturally, the PMS system must react immediately to new information at any time.At the end of the surveillance period, the collected data are summarized and evaluated in the corresponding report updates (PMS report / Periodic Safety Update Report (PSUR) as well as the PMCF report and the subsequent CER). If necessary, follow-up measures are defined in accordance with the PMS objectives. Plans do not usually need to be updated if the planning has proven to be complete and appropriate.Emergence of New RisksNew, changed, or emerging risks can become known at any time, in which case the PMS system must respond with the involvement of risk management (and other affected departments). In this case, it may be necessary to update the PMS plan in order to collect additional data. The new risk is added to the risk analysis and, if necessary, the Clinical Evaluation Plan (CEP) will also take into account the new risk as part of the planning of the clinical benefit-risk assessment. The additional measures for obtaining further necessary clinical data are defined in the PMCF plan.This can be done immediately and the ongoing activities are then expanded to include these new aspects. At the end of the data collection period, the relevant reports from PMS, risk management, and clinical evaluation are comprehensively updated.If changes are introduced to products with an existing MDR certification or to their intended purpose, these changes are treated as new developments. In this case, the corresponding additional or expanded plans, data, and reports must be generated. After a successful conformity assessment, the variant (or the successor model) can be placed on the market. The existing reports for the previous product will continue to be updated regularly until the end of its product life. It is of course possible to merge the documents.
PMS is carried out over the entire lifecycle (as defined by the manufacturer) of all products on the market, up to the last item on the market. This also applies to the reports that have to be updated (i.e. the PSUR). Therefore, the PMS system must remain active until the last moment when it is still possible to react, e.g. with a Field Safety Corrective Action (FSCA).During this time, you may collect enough clinical data from the PMCF activities to be able to gradually draw with certainty all the clinical conclusions necessary for the entire product life cycle. In this case, you can consider extending the update times for the Clinical Evaluation Report (CER). With an appropriate justification, you can write your last CER when product sales are stopped: a design change as a response to a problem would no longer take place after a sales stop and the PMS system remains active for all further reactive measures. However, the CER must be resumed if the benefit-risk ratio were to become unacceptable due to a new situation. The PMCF report is generally required for PMS and must therefore be continued. Continued PMCF activities are important to ensure the ability to react to unexpected clinical data.
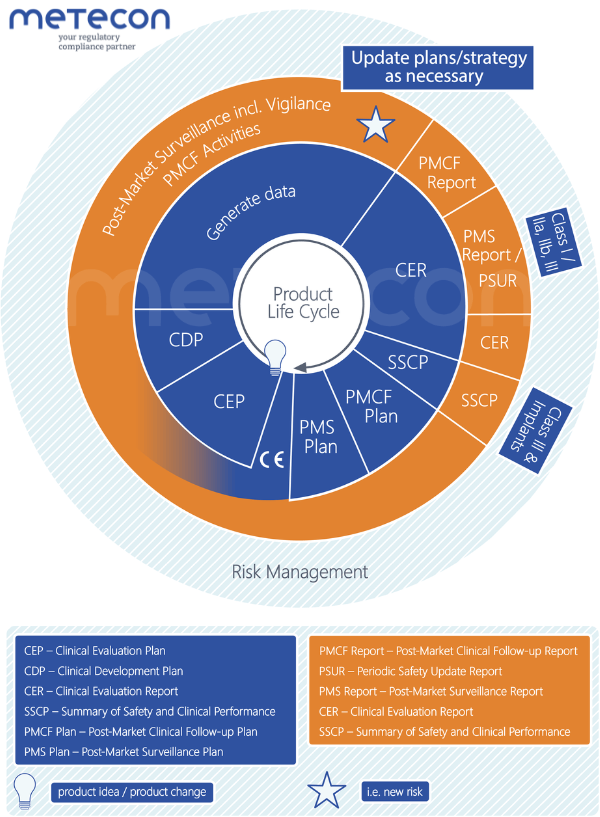
The following diagram shows the previously explained phases of the product lifecycle, i.e. development (blue), the post-marketing period (orange), as well as the plans and reports mentioned in their logical sequence - using the example of a newly developed product. This makes it easier to recognize the dependencies, which we will discuss below.
The Clinical Evaluation Plan defines the scope of the clinical evidence based on the relevant general safety and performance requirements (GSPR) in Annex I of the MDR. The CEP defines the aspects of the intended purpose (and thus the intended clinical performance and clinical benefit) and the residual risks according to the risk analysis to be analyzed. The parameters required to assess the benefit-risk ratio are identified. All marketing claims must also be substantiated by appropriate data and may thus require additional parameters.The strategy (route) of the clinical evaluation is determined taking into account these aspects and parameters, the preclinical and clinical data already available, the risk class, and possible equivalent products.The CEP is updated if the necessary scope or route changes.Finally, the search strategy used to collect the required data on the product is aligned with the parameters and the route. This strategy includes at least a literature search (with planning of sources, search terms, and selection as well as appraisal criteria) and, where necessary, a clinical study. An additional separate search strategy is set up to establish the state of the art (SOTA), in particular to identify the established expectation for benefits and performance and the accepted risks.
The clinical development plan (CDP) is formally part of the CEP. At the beginning of the development process, it outlines how you will generate the required clinical data. This can include exploratory studies, first-in-man studies, feasibility studies, and pilot studies up to proof-of-concept studies. In addition, an outlook on possible PMCF activities is also possible at this point.The CDP determines the objectives and endpoints of the study or studies based on the parameters to be evaluated. It also specifies acceptance criteria, milestones, a possible study design, and the time frame. The CDP is a structured umbrella document while the detailed planning of each study can be found in the respective Clinical Investigation Protocol (CIP).The CDP is updated when study objectives are changed or additional studies are added.
The Clinical Evaluation Report (CER) presents the results of the evaluation of all clinical data. Data from the product or from proven equivalent products are collected, selected, evaluated, and analyzed. It is examined whether and to what extent the requirements for performance/benefit and safety (risks and undesirable side effects) are met. Subsequently, the acceptability of the resulting benefit-risk ratio is assessed. Sufficient confirmatory clinical data are necessary for a conclusion on compliance with the GSPRs. Finally, the CER identifies the need for further clinical data to be collected as part of PMCF.The CER is continuously and regularly updated with PMCF data to assess the continued conformity with the general safety and performance requirements.
Post-Market Clinical Follow-up (PMCF) is formally part of PMS and is intended to extend the clinical evidence on the product over its entire lifecycle. PMCF also answers open questions that could previously only be estimated as part of the clinical evaluation (e.g. long-term behavior, monitoring of side effects and contraindications). The clinical evaluation is continuously and regularly updated with these data.In accordance with MDCG 2020-7, the PMCF plan describes the methods and procedures for the proactive collection or generation of clinical data. It elaborates activities that both seek additional data on selected parameters and support general monitoring and confirmation of use, performance and safety. The scope may vary depending on the PMCF activities required. A PMCF master plan can therefore be created at this point, which refers to various other plans in which the individual activities are defined in detail.The PMCF plan is updated when necessary, such as when the need for a change is identified in the PMCF report or when the CER has identified changed or additional PMCF requirements.
In the Post-Market Surveillance Plan, the manufacturer defines how the device is to be monitored after it has been placed on the market including the data that are to be collected. In the device-specific planning of these activities, the characteristics and associated risks of the device and the findings from the clinical evaluation report must be taken into account. Furthermore, the methods used to evaluate the data and the evaluation criteria are defined. Data collection on the market must be carried out proactively by the manufacturer. It is important that the planning is done in such a way that the PMS objectives from Article 83 of the MDR can be fulfilled.The PMS plan must be updated if there are new aspects to be considered (e.g. a new clinical benefit), if opportunities for improvement are identified in the current planning, or if there is a change in the devices covered by the PMS plan. The PMS plan also needs to be updated when new data sets become available that should be included in PMS. There are other situations where formal changes to the PMS plan are required.Proactive PMSWhen planning proactive PMS activities, it is important to consider which activities are to be carried out as PMCF activities. This is important as the PMCF activities are planned according to Annex XIV of the MDR, should meet the PMCF objectives, and are summarized in a PMCF report. If an activity is conducted as part of PMCF, then the data collected should meet the definition of clinical data according to Article 2(48) of the MDR. This means that not every proactive activity conducted as part of the PMS will meet the criteria for PMCF. The differentiation between a proactive PMS activity and a PMCF activity should be governed in the appropriate process. One sign that this was not done correctly is when the collected PMCF data have no clinical relevance.Typical proactive PMS or PMCF datasets are searches of the scientific literature and published reports on the clinical experience of either your own device or equivalent devices. Information regarding similar devices is also collected as part of an adverse event or field safety corrective actions (FSCAs) database search, often using the Manufacturer and User Facility Device Experience (MAUDE) database of the US Food & Drug Administration (FDA). User surveys can be conducted as either a proactive PMS or PMCF activity, depending on the survey objective.It is important that a user survey in the context of PMS/PMCF is properly planned and executed, i.e. from the objective of the survey to the data analysis and evaluation. If a user survey is supported by software, this software should also be suitable for this purpose. Several issues need to be considered here, such as data fields (free text or selection), language, data security, and features for the data evaluation.Reactive PMSAs a rule, reactive datasets are included in the PMS plan and not in the PMCF plan and include several of the datasets mentioned in Annex III of the MDR. These datasets include, for example, serious incidents, FSCAs, and trend reports in accordance with Article 88 of the MDR. As part of PMS, information and events are evaluated as soon as they are received and the corresponding follow-up measures are initiated immediately. In this case, the data contained in the report is a summary of the events during the surveillance period, including any necessary follow-up actions.In the PMS plan, the required information should be planned according to MDCG 2022-21 so that the data can be properly presented in the reports. As with the proactive data, the PMS plan should also describe how the reactive data analysis and evaluation will be carried out.
The clinical data and, if applicable, interim results from the PMCF activities on your device along with equivalent and / or similar devices are summarized and presented in accordance with MDCG 2020-8. The data are evaluated as to whether they potentially confirm or limit the previous conclusions in the clinical evaluation or whether they instead represent new information in comparison with the previous evaluation. As a rule, the data are checked for quality and the activities for effectiveness. The PMCF report can also decide on follow-up actions.The PMCF report is updated regularly. The update times follow the data collection periods as set in PMS.
The PMS Report in accordance with Article 85 of the MDR is created for Class I devices and includes a summary of the results from the PMS data (including PMCF data) obtained during the data collection period. Based on these results, a conclusion is drawn, which is then input, for example, to the clinical evaluation . Additionally, corrective and preventive actions (CAPAs) are outlined and justified. The purpose of the PMS Report is to gain insights into the safety and performance of the device on the market over its entire lifecycle, which can be used for further product development and to ensure product safety. Maintaining the PMS system over the entire product lifecycle ensures that the device meets the requirements of the MDR at all times.The Periodic Safety Update Report (PSUR) is created for Class IIa, IIb, and III products and, like the PMS report, contains a summary of the results from the market data obtained during the most recent data collection period. Here too, a conclusion is drawn on the basis of these results, which is input for the clinical evaluation, and CAPAs are presented and justified. In addition to the topics included in the PMS report, the PSUR also contains the conclusions from the benefit-risk assessment, the main findings from PMCF, and the volume of sales of the device as well as further information, e.g. on the frequency of use.The aim of the PSUR, as stated in MDCG 2022-21, is to provide a general overview of the PMS activities, the collected data and their analyis along with a summary of all results and conclusions. When preparing the PSUR, it is very important to make a statement regarding the data validity in order to openly address limitations. Conclusions are also drawn about new (or emerging) risks or benefits and potential negative impacts on the benefit-risk assessment (see Annex I of MDCG 2022-21). The PMCF conclusions are an essential contribution to the ongoing confirmation of an acceptable benefit-risk profile, and emphasize the importance of properly integrating PMCF into PMS.The PSUR is regularly updated (also for legacy devices). The update times are defined by the device risk class. The PMS report (for Class I devices) is prepared as required and should be updated shortly before the clinical evaluation is to be updated.
The SSCP report must be prepared for Class III and implantable devices in accordance with Article 32 of the MDR and MDCG 2019-9 Rev.1. The purpose of this summary report is to present the device in the context of its use, to explain residual risks and any undesirable effects, warnings and precautions, and to present alternative options with regard to therapy or diagnosis.The SSCP will be referenced in the instructions for use or labeling and will be made available to the public via EUDAMED. The report must be written in such a way that it can also be understood by laypersons, if they are the intended users.Before publication, the report is validated by the notified body, which then makes the report available in EUDAMED.The SSCP is updated annually.
During the data collection period, all planned activities take place to collect data needed to assess the continued performance and safety of the device on the market. Towards the end of this period, the associated reporting begins. This phase should be kept compact so that none of the reports are unduly delayed. During the writing phase, the PMS system will of course remain active and plans should generally be changed as soon as a need is identified. There is no need to wait to supplement the current activities with additional PMS activities that are found to be necessary.We have presented a smooth process that considers the interaction points between the created documents. Within the scope of this blog, however, we cannot adress every individual scenario, especially as special situations can arise at any time, such as the identification of new risks as mentioned above, but also the introduction of new or updated standards, or at a point in time when it becomes evident that you have to improve your PMS and / or PMCF activities.Nevertheless, we recommend that you focus on achieving the synchronized rhythm of reports as described above and then maintain your reports promptly. As this can be a challenge, you may find that you need one or two iterations before this will run smoothly.
Our experience shows that there can be numerous special situations that make it difficult to achieve the desired synchronized rhythm for all reports. Is this the case for you? Our team can work with you to develop a strategy for your product lifecycle reporting that suits your device and your requirements and helps you to react confidently to unforeseen circumstances. Please reach out to us and find out how quickly and easily we can help you!
Phases of the product lifecycle under the MDR
Risk Management accompanies the entire product lifecycle from the product idea to the end of life of the last item on the market. It must identify all risks, establish acceptance criteria, and assess the residual risk remaining after risk control measures have been implemented. To do this, risk management needs validation data such as that determined by the clinical evaluation.The Post-Market Surveillance (PMS) system accompanies the product lifecycle from market entry. Among other things, it must continuously ensure that new, changed or emerging risks are identified at all times, and - if necessary – that it is adapted accordingly and urgent measures are initiated immediately. The latter may include product changes and field measures.
Development and legacy devices
From the first product idea and over the entire product development, the collection of clinical data must be planned. These data must then be generated and analyzed. Following the clinical evaluation report (CER), the PMCF (Post-Market Clinical Follow-up) plan is created. For CE marking, the PMS plan must also be created, and Class III products and implants additionally require an initial SSCP (Summary of Safety and Clinical Performance).For legacy devices, it is important to check that all documents are available before trying to achieve CE marking under the MDR. Not all reports were required under the Medical Device Directive (MDD - Directive 93/42/EEC on medical devices) nor during the transition phase, which is why they might not be available. This mainly concerns the SSCP – and often the clinical evaluation is too minimalistic. However, these reports must now exist and be compliant for certification under the MDR. The available data for legacy devices must be sufficient from the point of view of the MDR and in accordance with the Medical Device Coordination Group’s guidance MDCG 2020-6.As the requirements for a PMS system in accordance with the MDR, including vigilance, and its documentation already apply to legacy devices, these plans and reports should already be available for legacy devices when it is time for certification under the MDR.
Post-market surveillance under the MDR
After successful CE marking and market entry, the data collection period that you will need to follow is determined by the risk class of your device. You will have a data collection period of 1 year for Class IIb and III medical devices, 2 years for Class IIa devices, or until a report is needed (Class I). During this period, the planned PMS and PMCF activities are carried out, data are collected , and records created. Naturally, the PMS system must react immediately to new information at any time.At the end of the surveillance period, the collected data are summarized and evaluated in the corresponding report updates (PMS report / Periodic Safety Update Report (PSUR) as well as the PMCF report and the subsequent CER). If necessary, follow-up measures are defined in accordance with the PMS objectives. Plans do not usually need to be updated if the planning has proven to be complete and appropriate.Emergence of New RisksNew, changed, or emerging risks can become known at any time, in which case the PMS system must respond with the involvement of risk management (and other affected departments). In this case, it may be necessary to update the PMS plan in order to collect additional data. The new risk is added to the risk analysis and, if necessary, the Clinical Evaluation Plan (CEP) will also take into account the new risk as part of the planning of the clinical benefit-risk assessment. The additional measures for obtaining further necessary clinical data are defined in the PMCF plan.This can be done immediately and the ongoing activities are then expanded to include these new aspects. At the end of the data collection period, the relevant reports from PMS, risk management, and clinical evaluation are comprehensively updated.If changes are introduced to products with an existing MDR certification or to their intended purpose, these changes are treated as new developments. In this case, the corresponding additional or expanded plans, data, and reports must be generated. After a successful conformity assessment, the variant (or the successor model) can be placed on the market. The existing reports for the previous product will continue to be updated regularly until the end of its product life. It is of course possible to merge the documents.
End of product life
PMS is carried out over the entire lifecycle (as defined by the manufacturer) of all products on the market, up to the last item on the market. This also applies to the reports that have to be updated (i.e. the PSUR). Therefore, the PMS system must remain active until the last moment when it is still possible to react, e.g. with a Field Safety Corrective Action (FSCA).During this time, you may collect enough clinical data from the PMCF activities to be able to gradually draw with certainty all the clinical conclusions necessary for the entire product life cycle. In this case, you can consider extending the update times for the Clinical Evaluation Report (CER). With an appropriate justification, you can write your last CER when product sales are stopped: a design change as a response to a problem would no longer take place after a sales stop and the PMS system remains active for all further reactive measures. However, the CER must be resumed if the benefit-risk ratio were to become unacceptable due to a new situation. The PMCF report is generally required for PMS and must therefore be continued. Continued PMCF activities are important to ensure the ability to react to unexpected clinical data.
Plans and reports
The following diagram shows the previously explained phases of the product lifecycle, i.e. development (blue), the post-marketing period (orange), as well as the plans and reports mentioned in their logical sequence - using the example of a newly developed product. This makes it easier to recognize the dependencies, which we will discuss below.
Clinical evaluation plan (CEP)
The Clinical Evaluation Plan defines the scope of the clinical evidence based on the relevant general safety and performance requirements (GSPR) in Annex I of the MDR. The CEP defines the aspects of the intended purpose (and thus the intended clinical performance and clinical benefit) and the residual risks according to the risk analysis to be analyzed. The parameters required to assess the benefit-risk ratio are identified. All marketing claims must also be substantiated by appropriate data and may thus require additional parameters.The strategy (route) of the clinical evaluation is determined taking into account these aspects and parameters, the preclinical and clinical data already available, the risk class, and possible equivalent products.The CEP is updated if the necessary scope or route changes.Finally, the search strategy used to collect the required data on the product is aligned with the parameters and the route. This strategy includes at least a literature search (with planning of sources, search terms, and selection as well as appraisal criteria) and, where necessary, a clinical study. An additional separate search strategy is set up to establish the state of the art (SOTA), in particular to identify the established expectation for benefits and performance and the accepted risks.
Clinical development plan (CDP)
The clinical development plan (CDP) is formally part of the CEP. At the beginning of the development process, it outlines how you will generate the required clinical data. This can include exploratory studies, first-in-man studies, feasibility studies, and pilot studies up to proof-of-concept studies. In addition, an outlook on possible PMCF activities is also possible at this point.The CDP determines the objectives and endpoints of the study or studies based on the parameters to be evaluated. It also specifies acceptance criteria, milestones, a possible study design, and the time frame. The CDP is a structured umbrella document while the detailed planning of each study can be found in the respective Clinical Investigation Protocol (CIP).The CDP is updated when study objectives are changed or additional studies are added.
Clinical evaluation report (CER)
The Clinical Evaluation Report (CER) presents the results of the evaluation of all clinical data. Data from the product or from proven equivalent products are collected, selected, evaluated, and analyzed. It is examined whether and to what extent the requirements for performance/benefit and safety (risks and undesirable side effects) are met. Subsequently, the acceptability of the resulting benefit-risk ratio is assessed. Sufficient confirmatory clinical data are necessary for a conclusion on compliance with the GSPRs. Finally, the CER identifies the need for further clinical data to be collected as part of PMCF.The CER is continuously and regularly updated with PMCF data to assess the continued conformity with the general safety and performance requirements.
PMCF-Plan
Post-Market Clinical Follow-up (PMCF) is formally part of PMS and is intended to extend the clinical evidence on the product over its entire lifecycle. PMCF also answers open questions that could previously only be estimated as part of the clinical evaluation (e.g. long-term behavior, monitoring of side effects and contraindications). The clinical evaluation is continuously and regularly updated with these data.In accordance with MDCG 2020-7, the PMCF plan describes the methods and procedures for the proactive collection or generation of clinical data. It elaborates activities that both seek additional data on selected parameters and support general monitoring and confirmation of use, performance and safety. The scope may vary depending on the PMCF activities required. A PMCF master plan can therefore be created at this point, which refers to various other plans in which the individual activities are defined in detail.The PMCF plan is updated when necessary, such as when the need for a change is identified in the PMCF report or when the CER has identified changed or additional PMCF requirements.
PMS Plan
In the Post-Market Surveillance Plan, the manufacturer defines how the device is to be monitored after it has been placed on the market including the data that are to be collected. In the device-specific planning of these activities, the characteristics and associated risks of the device and the findings from the clinical evaluation report must be taken into account. Furthermore, the methods used to evaluate the data and the evaluation criteria are defined. Data collection on the market must be carried out proactively by the manufacturer. It is important that the planning is done in such a way that the PMS objectives from Article 83 of the MDR can be fulfilled.The PMS plan must be updated if there are new aspects to be considered (e.g. a new clinical benefit), if opportunities for improvement are identified in the current planning, or if there is a change in the devices covered by the PMS plan. The PMS plan also needs to be updated when new data sets become available that should be included in PMS. There are other situations where formal changes to the PMS plan are required.Proactive PMSWhen planning proactive PMS activities, it is important to consider which activities are to be carried out as PMCF activities. This is important as the PMCF activities are planned according to Annex XIV of the MDR, should meet the PMCF objectives, and are summarized in a PMCF report. If an activity is conducted as part of PMCF, then the data collected should meet the definition of clinical data according to Article 2(48) of the MDR. This means that not every proactive activity conducted as part of the PMS will meet the criteria for PMCF. The differentiation between a proactive PMS activity and a PMCF activity should be governed in the appropriate process. One sign that this was not done correctly is when the collected PMCF data have no clinical relevance.Typical proactive PMS or PMCF datasets are searches of the scientific literature and published reports on the clinical experience of either your own device or equivalent devices. Information regarding similar devices is also collected as part of an adverse event or field safety corrective actions (FSCAs) database search, often using the Manufacturer and User Facility Device Experience (MAUDE) database of the US Food & Drug Administration (FDA). User surveys can be conducted as either a proactive PMS or PMCF activity, depending on the survey objective.It is important that a user survey in the context of PMS/PMCF is properly planned and executed, i.e. from the objective of the survey to the data analysis and evaluation. If a user survey is supported by software, this software should also be suitable for this purpose. Several issues need to be considered here, such as data fields (free text or selection), language, data security, and features for the data evaluation.Reactive PMSAs a rule, reactive datasets are included in the PMS plan and not in the PMCF plan and include several of the datasets mentioned in Annex III of the MDR. These datasets include, for example, serious incidents, FSCAs, and trend reports in accordance with Article 88 of the MDR. As part of PMS, information and events are evaluated as soon as they are received and the corresponding follow-up measures are initiated immediately. In this case, the data contained in the report is a summary of the events during the surveillance period, including any necessary follow-up actions.In the PMS plan, the required information should be planned according to MDCG 2022-21 so that the data can be properly presented in the reports. As with the proactive data, the PMS plan should also describe how the reactive data analysis and evaluation will be carried out.
PMCF Report
The clinical data and, if applicable, interim results from the PMCF activities on your device along with equivalent and / or similar devices are summarized and presented in accordance with MDCG 2020-8. The data are evaluated as to whether they potentially confirm or limit the previous conclusions in the clinical evaluation or whether they instead represent new information in comparison with the previous evaluation. As a rule, the data are checked for quality and the activities for effectiveness. The PMCF report can also decide on follow-up actions.The PMCF report is updated regularly. The update times follow the data collection periods as set in PMS.
PMS Report / PSUR
The PMS Report in accordance with Article 85 of the MDR is created for Class I devices and includes a summary of the results from the PMS data (including PMCF data) obtained during the data collection period. Based on these results, a conclusion is drawn, which is then input, for example, to the clinical evaluation . Additionally, corrective and preventive actions (CAPAs) are outlined and justified. The purpose of the PMS Report is to gain insights into the safety and performance of the device on the market over its entire lifecycle, which can be used for further product development and to ensure product safety. Maintaining the PMS system over the entire product lifecycle ensures that the device meets the requirements of the MDR at all times.The Periodic Safety Update Report (PSUR) is created for Class IIa, IIb, and III products and, like the PMS report, contains a summary of the results from the market data obtained during the most recent data collection period. Here too, a conclusion is drawn on the basis of these results, which is input for the clinical evaluation, and CAPAs are presented and justified. In addition to the topics included in the PMS report, the PSUR also contains the conclusions from the benefit-risk assessment, the main findings from PMCF, and the volume of sales of the device as well as further information, e.g. on the frequency of use.The aim of the PSUR, as stated in MDCG 2022-21, is to provide a general overview of the PMS activities, the collected data and their analyis along with a summary of all results and conclusions. When preparing the PSUR, it is very important to make a statement regarding the data validity in order to openly address limitations. Conclusions are also drawn about new (or emerging) risks or benefits and potential negative impacts on the benefit-risk assessment (see Annex I of MDCG 2022-21). The PMCF conclusions are an essential contribution to the ongoing confirmation of an acceptable benefit-risk profile, and emphasize the importance of properly integrating PMCF into PMS.The PSUR is regularly updated (also for legacy devices). The update times are defined by the device risk class. The PMS report (for Class I devices) is prepared as required and should be updated shortly before the clinical evaluation is to be updated.
Summary of safety and clinical performance (SSCP)
The SSCP report must be prepared for Class III and implantable devices in accordance with Article 32 of the MDR and MDCG 2019-9 Rev.1. The purpose of this summary report is to present the device in the context of its use, to explain residual risks and any undesirable effects, warnings and precautions, and to present alternative options with regard to therapy or diagnosis.The SSCP will be referenced in the instructions for use or labeling and will be made available to the public via EUDAMED. The report must be written in such a way that it can also be understood by laypersons, if they are the intended users.Before publication, the report is validated by the notified body, which then makes the report available in EUDAMED.The SSCP is updated annually.
Conclusion
During the data collection period, all planned activities take place to collect data needed to assess the continued performance and safety of the device on the market. Towards the end of this period, the associated reporting begins. This phase should be kept compact so that none of the reports are unduly delayed. During the writing phase, the PMS system will of course remain active and plans should generally be changed as soon as a need is identified. There is no need to wait to supplement the current activities with additional PMS activities that are found to be necessary.We have presented a smooth process that considers the interaction points between the created documents. Within the scope of this blog, however, we cannot adress every individual scenario, especially as special situations can arise at any time, such as the identification of new risks as mentioned above, but also the introduction of new or updated standards, or at a point in time when it becomes evident that you have to improve your PMS and / or PMCF activities.Nevertheless, we recommend that you focus on achieving the synchronized rhythm of reports as described above and then maintain your reports promptly. As this can be a challenge, you may find that you need one or two iterations before this will run smoothly.
How can we help?
Our experience shows that there can be numerous special situations that make it difficult to achieve the desired synchronized rhythm for all reports. Is this the case for you? Our team can work with you to develop a strategy for your product lifecycle reporting that suits your device and your requirements and helps you to react confidently to unforeseen circumstances. Please reach out to us and find out how quickly and easily we can help you!
Our blog posts are researched and created with the utmost care, but are only snapshots of the regulations, which are constantly changing. We do not guarantee that older content is still current or meaningful. If you are not sure whether the article you have read on this page still corresponds to the current state of regulation, please contact us: we will quickly place your topic in the current context.





