
MDR-Praxis, Teil V: Die Dokumente Ihrer Technischen Dokumentation
28/10/2019
Do you have any questions about the article or would you like to find out more about our services? We look forward to hearing from you!Make a non-binding enquiry now
From our archives (28/10/2019)
Willkommen zum 5. Teil unserer Reihe „MDR-Praxis“! Teil 1-3 finden Sie als Download hier, Teil 4 in unseren News. Unser heutiges Thema: Welche Dokumente entstehen eigentlich konkret bei der Überarbeitung bzw. Erstellung Ihrer Technischen Dokumentation? Die MDR gibt Ihnen zwar nicht vor, wie Sie Ihre Dokumente zusammenstellen und in welchen Dokumenten Sie welche Inhalte wiedergeben sollen. Aber die MDR fordert in Anhang II verschiedene Inhalte von Ihnen als Hersteller, mit denen Sie die Konformität Ihres Produkts gegenüber den Anforderungen der Verordnung in Anhang I, den Grundlegenden Sicherheits- und Leistungsanforderungen, nachweisen; s. hierzu auch Artikel 10, Allgemeine Pflichten der Hersteller:„(4) Die Hersteller von Produkten, bei denen es sich nicht um Sonderanfertigungen handelt, verfassen eine technische Dokumentation für diese Produkte und halten diese Dokumentation auf dem neuesten Stand. Die technische Dokumentation ist so beschaffen, dass durch sie eine Bewertung der Konformität des Produkts mit den Anforderungen dieser Verordnung ermöglicht wird. Die technische Dokumentation enthält die in den Anhängen II und III aufgeführten Elemente.“Ohne Abstimmung und Struktur geht es nicht
Bevor Sie jetzt jedoch sofort loslegen: Stimmen Sie sich hierzu unbedingt mit Ihrer Benannten Stelle ab! Es gibt auf Europäischer Ebene momentan zwar keine einheitlichen Vorgaben, aber die Benannten Stellen haben im Rahmen ihrer eigenen Umsetzung der MDR durchaus Strukturen für die Technische Dokumentation definiert und erwarten von ihren Kunden einen entsprechenden Aufbau der Technischen Dokumentation.Warum Benannte Stellen diese Strukturen vorgeben? Weil es ohne sie unmöglich ist, die bevorstehenden Produktaktenprüfungen in einer halbwegs annehmbaren Zeit zu organisieren und durchzuführen. Schließlich ist es nicht nur in Ihrem Interesse als Hersteller, sondern auch im Interesse Ihrer Benannten Stelle, Ihre Dokumentation schnellstmöglich zu prüfen und Ihnen somit einen möglichst guten Marktzugang unter der MDR zu ermöglichen. Und auch im Hinblick auf die Anforderungen an die Eigenschaften der Technischen Dokumentation macht ein strukturiertes Vorgehen Sinn, denn - wir erinnern uns – die Form der Technischen Dokumentation soll klar, organisiert, leicht durchsuchbar und eindeutig sein.
Tipps für die Erstellung und Pflege der Technischen Dokumentation
- Vermeiden Sie redundante Informationen.
- Es gibt gewisse, besonders wichtige Dokumente, die führend sind und als Basis oder Ursprung die Ausgangsinformationen bereitstellen, die in anderen Dokumenten wiedergegeben werden.
- Wo es sinnvoll ist, können und sollten Sie mit Verweisen arbeiten.
- Sofern Dokumente zum Produktverständnis Informationen aus oben genannten „Ursprungsdokumenten“ aufgreifen und inhaltlich wiedergeben (da sie als Stand-alone-Dokumente zu sehen sind), sollten diese Inhalte übernommen werden und nicht mit gegebenenfalls anderem Wortlaut neu geschrieben werden.
Ein Punkt, der mir besonders am Herzen liegt, und dessen Herausforderung mir in der täglichen Arbeit immer wieder bewusst wird, ist die Notwendigkeit, die Kompetenz Ihrer Fachkollegen für die Erstellung eines Dokuments zu nutzen. Die Fachabteilungen und Regulatory Affairs sollten gemeinsam an der Erstellung eines Dokuments arbeiten! Die Gründe hierfür liegen auf der Hand:
Die Verordnung stellt viele regulatorische Anforderungen und verschiedene Begriffe sind mit Definitionen belegt, die die Techniker, Entwickler und Anwender nicht immer kennen - und dies im Übrigen auch nicht müssen.
- Jedes Dokument hat eine andere Intension, einen anderen Adressaten, für den es die Informationen bereithält.
- Manche Dokumente sind als Stand-alone-Dokumente zu sehen (Klinische Bewertung, Biologische Bewertung).
Deshalb meine Empfehlung: Nutzen Sie die Kenntnisse aller Kollegen und erstellen Sie dann die Dokumente mit der entsprechenden Zielstellung, die dieses Dokument hat.Ein Beispiel: Die Produktbeschreibung
Hierbei handelt es sich um ein regulatorisches Dokument, dass das Produkt mit seinen wichtigsten Eigenschaften beschreiben soll. Es dient vor allem Dritten dafür, ein Verständnis für das Produkt zu bekommen und es in seiner Auslegung und seiner generellen Anwendung zu verstehen. Schreibt der Mediziner dieses Dokument, liest es sich deutlich anders, als wenn der RA-Manager dieses Dokument auf Basis des Inputs der Entwicklung und der Kliniker/Anwender (aus der Gebrauchsanweisung) erstellt.
Anders sieht es dagegen bei der Erstellung der Gebrauchsanweisung aus. Diese ist für den Anwender bestimmt, und sollte dementsprechend die Informationen für die Zielgruppe derart bereitstellen, dass der Anwender diese versteht.Schauen wir jetzt also gemeinsam in die Inhalte und fassen die wichtigsten Punkte zusammen:
Überarbeitung der Technischen Dokumentation: Welche Dokumente entstehen?
Die nachfolgenden Ausführungen sollen Ihnen einen Überblick geben, worauf es bei den in Anhang II aufgeführten Bestandteilen der Technischen Dokumentation ankommt. Ebenfalls benenne ich mögliche Dokumente, in denen diese Inhalte sinnvollerweise dargelegt werden. Bitte beachten Sie, dass dies nur Vorschläge sind. Der Aufbau Ihrer Technischen Dokumentation kann durchaus davon abweichen.
1. PRODUKTBESCHREIBUNG UND SPEZIFIKATION, EINSCHLIESSLICH DER VARIANTEN UND ZUBEHÖRTEILE
1.1 Produktbeschreibung und SpezifikationenWorauf kommt es an?
Dieses Kapitel dient der eindeutigen Identifizierung und Beschreibung des Produkts. Es soll klar dargestellt werden, in welcher Form das Produkt wirkt und was seine Funktionselemente sind, mit denen es seine Leistung erbringt. Dies setzt voraus, dass die Zweckbestimmung formuliert ist. Auf Basis der Zweckbestimmung können Sie entscheiden, ob es sich bei Ihrem Produkt per Definition um ein Medizinprodukt im Sinne der Verordnung handelt oder um ein Zubehör, und dies entsprechend begründen.
Bitte beachten Sie, dass es keinen Unterschied macht, ob es sich um ein Medizinprodukt oder um Zubehör handelt: Auch Zubehör muss die Grundlegenden Sicherheits- und Leistungsanforderungen erfüllen und wird wie das eigentliche Medizinprodukt behandelt und dokumentiert.Mögliche Dokumente:
- Name und Anschrift des Herstellers,
- Zweckbestimmung inklusive bestimmungsgemäßer Verwendung:
- Intended Purpose,
- Intended Use,
- Produktbeschreibung:
- Allgemeine Produktbeschreibung,
- Darstellung der Varianten,
- Übersicht über Produkte in den Produktgruppen/Gerätetypen (z. B. tabellarische Darstellung inklusive technischer Spezifikationen),
- Benennung des Zubehörs,
- Verwendung mit anderen Produkten,
- Anwendungsbereiche,
- Indikationen,
- Kontraindikationen,
- Beschreibung der Funktionsweise und Benennung der Funktionselemente,
- Produktklassifizierung:
- Begründung Medizinprodukt,
- Nachweis der Klassifizierungsregeln,
- UDI,
- Konformitätserklärung,
- Kurzbericht über Sicherheit und klinische Leistung (nur implantierbare Medizinprodukte und Produkte der Klasse III),
- Erklärung zu besonderen Substanzen,
- Übersicht über Rohstoffe, Komponenten und Packmittel.
1.2 Hinweis auf frühere und ähnliche Generationen des ProduktsWorauf kommt es an?
Die Verordnung fordert in erster Linie eine Übersicht über die eigenen früheren Produkte bzw. über ähnliche Produkte sowie Wettbewerbsprodukte, die sich am Markt befinden, d. h. eine tabellarische Auflistung von Vorgängerversionen Ihres Produkts sollte diese Anforderung erst einmal ausreichend belegen. Wenn Sie ein wenig mehr liefern wollen, können Sie die Unterschiede zwischen den Produkten noch benennen.
Die Informationen hierzu sollte Ihnen Ihre Änderungshistorie gemäß EN ISO 13485 Punkt 7.3.9 „Lenkung von Entwicklungsänderungen“ liefern können. Sprechen Sie mit Ihren Kollegen aus QM, wie die Anforderungen der EN ISO 13485 in den Punkten 4.2.3 „Medizinprodukteakte“ und 7.3.9 „Lenkung von Entwicklungsänderungen“ in Verbindung mit 7.3.10 „Entwicklungsakten“ bisher umgesetzt sind.Bei den Wettbewerbsprodukten gilt es, nicht nur den europäischen CE-Raum zu recherchieren, sondern auch internationale Märkte zu berücksichtigen.Mögliche Dokumente:
- Produktbeschreibung – mindestens als eigenes Kapitel im Dokument, gegebenenfalls aber auch als eigenes Dokument,
- frühere und ähnliche Generationen des Produkts.
2. VOM HERSTELLER ZU LIEFERNDE INFORMATIONEN
Worauf kommt es an?
Mit diesen Informationen teilen Sie dem Anwender alles mit, was er zur sicheren Identifizierung und Anwendung Ihres Produkts wissen muss. Die Angaben entsprechen der Verwendung des Produkts im Sinne seiner Zweckbestimmung.
Die Inhalte der Gebrauchsanweisung richten sich speziell an den Anwender. Dies kann ganz verschiedene Personengruppen der Gesundheitsberufe einschließen, die das Produkt anwenden. Beachten Sie, dass bei einigen Produkten auch „Laien“ mit dem Produkt umgehen und in diesem Fall die Anwender sind. Entsprechend verständlich müssen die Inhalte formuliert sein.
Die Gebrauchsanweisung kann auch Informationen für Dritte enthalten, wenn es z. B. um die Wiederaufbereitung des Produkts geht oder um spezielle Wartungs- und Pflegehinweise.Die Anforderungen an die Gebrauchsanweisung sind in Anhang I, 23.4 spezifiziert. Die Inhalte kommen aus verschiedenen Quellen, die im Laufe der Produktentwicklung entstehen, und werden in der Gebrauchsanweisung zusammengefasst. Jegliche Warn- und Sicherheitshinweise sollten als Maßnahme aus der Risikoanalyse kommen. Auch die CE-Kennzeichnung ist gemäß Artikel 20 Bestandteil Ihres Produkts.Die Dokumente einschließlich der Angaben auf der Transportverpackung sind in den Sprachen vorzuhalten, in denen das Produkt verkauft wird und die von den Mitgliedsstaaten akzeptiert werden. Dies gilt ebenfalls für Angaben auf der Transportverpackung, sofern es sich um relevante Informationen handelt, die erforderlich sind, um die Sicherheit und Leistungsfähigkeit des Produkts zu erhalten.Eine Empfehlung möchte ich Ihnen im Hinblick auf Klarheit und Eindeutigkeit in Ihren Dokumenten noch geben: Beachten Sie Artikel 7 „Angaben“ der MDR und prüfen Sie, dass die von Ihnen gemachten Informationen in den Dokumenten den tatsächlichen Produktspezifikationen und Eigenschaften Ihres Medizinprodukts entsprechen. Es dürfen keine widersprüchlichen und irreführenden Angaben gemacht werden, die den Anwender hinsichtlich der Zweckbestimmung, der Sicherheit und der Leistung des Produkts im Unklaren lassen, oder über die tatsächlichen Eigenschaften des Produkts hinausgehen.
Dies schließt Marketing und Werbebroschüren unbedingt mit ein!Mögliche Dokumente:
- Kennzeichnung Produkt,
- Kennzeichnung Produktverpackung,
- Kennzeichnung Transportverpackung,
- Gebrauchsanweisung,
- Wiederaufbereitungsanweisung (kann Bestandteil der Gebrauchsanweisung sein),
- Installations- und Wartungsanweisungen.
3. INFORMATIONEN ZU AUSLEGUNG UND HERSTELLUNG
Worauf kommt es an?
Die einzelnen Phasen der Produktauslegung sollen nachvollziehbar dargestellt sein. Die erwarteten Informationen gehen hier mit Sicherheit über die Anforderungen der MDD hinaus. Auch hier sollte Ihnen Ihr QMS Input liefern können, vorrangig über die Prozesse zur Planung der Produktrealisierung und der Entwicklungsplanung.
Die Informationen und Spezifikationen sind vollständig in der Technischen Dokumentation aufzunehmen. Dies schließt Informationen zu den Herstellprozessen und deren Validierung mit ein.
Neu gefordert sind Angaben zur Qualitätssicherung des Produkts.
Wichtig ist, dass die Angaben alle Standorte, inklusive ausgelagerte Prozesse bei Lieferanten und Unterauftragnehmer, einschließen, die an der Entwicklung und Produktion des Produkts beteiligt sind.Mögliche Dokumente:
- Beschreibung der Auslegung:
- Beschreibung des Entwicklungsprozesses,
- Darstellung der Auslegungsphasen,
- Ergebnisse der einzelnen Meilensteine,
- Benennung aller beteiligten Stellen, die in der Auslegungsphase involviert waren (Entwicklungspartner, Forschungseinrichtungen, etc.),
- Beschreibung der Produktion:
- Überblick über die Produktion, dies kann gut in Form von Flussdiagrammen erfolgen und Informationen wie eingehende und ausgehende Produktbestandteile/ Komponenten und Dokumente einschließen,
- Übersicht der Produktionsstätten,
- Benennung spezieller Prozesse inklusive deren Validierung,
- Arbeitspläne,
- Prüfpläne,
- Informationen zu kontrollierten Bedingungen, die für gewisse Herstellschritte eingehalten werden müssen,
- Beschreibung der Qualitätskontrolle:
- Darlegung der einzelnen Prüfschritte (Inprozess und Endprüfungen) und deren Dokumentation.
4. GRUNDLEGENDE SICHERHEITS- UND LEISTUNGSANFORDERUNGEN
Worauf kommt es an?
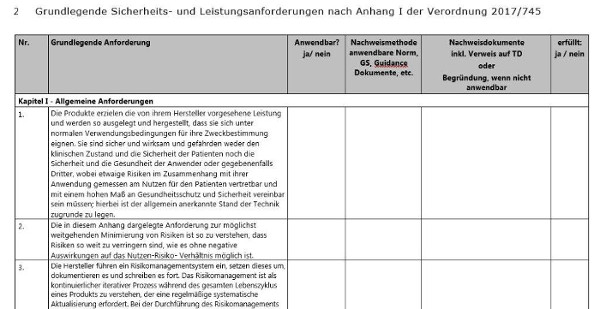
Sie müssen in einer systematischen Weise nachweisen, welche Anforderungen aus Anhang I auf Ihr Medizinprodukt zutreffen, und begründen, wenn bestimmte Anforderungen nicht zutreffen und wie Sie diese erfüllen und mit Nachweisdokumenten belegen.Wie bereits in Teil II dieser Reihe vorgeschlagen, empfiehlt sich eine Checkliste, die alle geforderten Punkte berücksichtigt:
- Begründung der Anwendbarkeit oder Nichtanwendbarkeit der jeweiligen Anforderung,
- Verweis auf angewandte Gemeinsame Spezifikationen und Normen (auch, wenn diese nur teilweise angewendet werden),
- Der Ausgabestand der Norm muss nachvollziehbar sein, kann aber auch in einer separaten Normenliste geführt werden,
- Eindeutiger Verweis auf gelenkte Dokumente und Aufzeichnungen zum Nachweis der Erfüllung,
- Bewertung, ob die Anforderungen erfüllt sind.
 Klicken Sie hier für eine größere Ansicht
Klicken Sie hier für eine größere AnsichtMögliche Dokumente:
- Checkliste der Erfüllung der Grundlegenden Sicherheits- und Leistungsanforderungen,
- Liste der angewandten Normen und (wenn vorhanden) der gemeinsamen Spezifikationen.
5. NUTZEN-RISIKO-ANALYSE UND RISIKOMANAGEMENT
Worauf kommt es an?
Wie die Überschrift schon zeigt, muss hier neben der eigentlichen Risikoanalyse das gesamte Risikomanagement dargelegt werden: Was ist für Ihr Produkt geplant? Welche Risiken werden identifiziert? Welche Maßnahmen ergreifen Sie, um die Risiken zu reduzieren?Dem Risikomanagement kommt eine besondere Bedeutung zu, da in den vergangenen Jahren immer mehr Normen auf den risikobasierten Ansatz zurückgreifen. Das bedeutet für Sie als Hersteller: Sie definieren das Risiko Ihres Produkts unter Berücksichtigung seiner Zweckbestimmung und seiner bestimmungsgemäßen Verwendung. Dies kann maßgeblichen Einfluss haben auf Verifikationstätigkeiten, z. B. nach EN ISO 60601-1 oder EN ISO 60601-1-2. Mögliche Dokumente:
- Risikomanagementplan,
- Risikoanalyse inklusive Risikokontrollmaßnahmen,
- Risikomanagementbericht einschließlich Bewertung der Restrisiken und des Risiko-Nutzen-Verhältnisses.
6. VERIFIZIERUNG UND VALIDIERUNG DES PRODUKTS
Worauf kommt es an?
Der Inhalt dieses Kapitels richtet sich nach den Spezifikationen Ihres Produkts und seinen Eigenschaften. Da in den meisten Fällen diverse einzelne Prüfungen zu den Themen durchgeführt werden, bietet sich auch hier eine übergreifende und zusammenfassende bewertende Dokumentation an.Mögliche Strukturen für den Aufbau
- Verifikationen:
- Gesamtprüfplan Verifikation,
- Prüfpläne und Prüfprotokolle:
- Biokompatibilität,
- Mechanische Prüfungen,
- Physikalische, chemische und mikrobiologische Prüfungen,
- Weitere präklinische Prüfungen,
- etc.
- Validierungen:
- Gebrauchstauglichkeit,
- Klinische Bewertung,
- etc.
Genauso können Sie die Dokumentation nach den Eigenschaften des Produkts strukturieren:
- Biokompatibilität: Berücksichtigung aller Materialien und Komponenten, welche einen direkten oder indirekten Kontakt zum Patienten oder Anwender haben können:
- Chemische Charakterisierung der Materialien,
- Literaturrecherchen,
- Prüfberichte zu durchgeführten biologischen Prüfungen.
Zusammenfassende Bewertung aller Daten und Prüfergebnisse für das Gesamtprodukt.
- Physikalische, chemische und mikrobiologische Prüfungen: Nachweise der Charakterisierung und präklinische Eignung der Produkte hinsichtlich anwendbarer Prüfparameter (z. B. physikalische Zusammensetzung, chemische Charakterisierung und Reinheit von Rohstoffen und Fertigprodukt, Mikrobiologischer Zustand des Endproduktes etc.):
- Planung und Übersicht über durchgeführte Prüfungen,
- Prüfberichte zu durchgeführten Prüfungen.
Zusammenfassende Bewertung aller Daten und Prüfergebnisse für das Gesamtprodukt.
- Elektrische Sicherheit und EMV (wenn zutreffend):
- Planung und Übersicht über durchgeführte Prüfungen,
- Prüfberichte zu durchgeführten Prüfungen,
- Bewertung der Daten und Prüfergebnisse.
- Software Verifizierung und Validierung (wenn zutreffend):
- Beschreibung des Softwarelebenszyklus,
- Beschreibung des Softwaredesigns,
- Validierung der Software, wie sie im fertigen Produkt verwendet wird.
Blick in die Zukunft für Ihr nächstes Medizinprodukt
Die nachfolgenden Überlegungen sind prospektiv und dienen dazu, für die zukünftige Entwicklung eines Medizinprodukts eine solide Basis in Ihrem QM-System zu schaffen. Die Technische Dokumentation fällt am Ende der Entwicklung nicht vom Himmel. Auch wird in der Regel keiner an Ihrem Schreibtisch vorbeikommen und Ihnen eine fertige Akte auf den Tisch oder vielmehr in den elektronischen Ordner legen. Und wenn doch, dann kümmern Sie sich gut um diesen Kollegen, denn der ist im wahrsten Sinne des Wortes unbezahlbar … :-) Die Technische Dokumentation bzw. die notwendigen Inhalte zur Erstellung der Dokumente entstehen gemeinsam mit ihrem Produkt, im Rahmen der Entwicklung. Es macht also Sinn, die „Dokumentenentstehung“ in Ihre Prozesse zu integrieren.Dabei helfen die folgenden Fragestellungen:
- Welche Bedeutung hat der geforderte Inhalt im Produktlebenszyklus?
- Welche Dokumente repräsentieren typischerweise den geforderten Inhalt?
- Aus welchen Quellen könnte der geforderte Inhalt entnommen werden?
- Wann sollte mit der Erstellung des geforderten Inhalts begonnen werden?
- Wann sollte (oder muss) der geforderte Inhalt (spätestens) vorliegen?
Ich wünsche Ihnen viel Erfolg bei der Überarbeitung oder der Neuerstellung Ihrer Dokumente. Und ich hoffe, dass die MDR Sie und Ihre Kollegen an einen Tisch bringt und Sie sich austauschen und gemeinsam Lösungen zur Umsetzung finden, diskutieren und implementieren.Nächstes Mal dreht sich alles um die Technische Dokumentation nach dem Inverkehrbringen Ihres Produkts. Die Technische Dokumentation sollte so aktuell sein wie Ihre Produkte. Erkenntnisse vom Markt fließen ins Unternehmen zurück, werden bewertet und haben gegebenenfalls einen Einfluss auf Ihr Produkt. Jegliche Änderungen sind in der Technischen Dokumentation abzubilden.
Trigger gibt es da verschiedene:
- Produktänderungen,
- geänderte regulatorische Anforderungen oder Normen zur Konformitätsvermutung,
- zeitliche regulatorische Vorgaben, die Sie einhalten müssen (Beispiel Berichte).
Our blog posts are researched and created with the utmost care, but are only snapshots of the regulations, which are constantly changing. We do not guarantee that older content is still current or meaningful. If you are not sure whether the article you have read on this page still corresponds to the current state of regulation, please contact us: we will quickly place your topic in the current context.




{kind=link}
