
A Guide to Process Validation - Smooth sailing, not stormy seas
07/05/2024
Do you have any questions about the article or would you like to find out more about our services? We look forward to hearing from you!Make a non-binding enquiry now
A deep dive into process validation: Where is validation necessary in production, and where are alternatives such as 100% verification of the result conceivable? In this article, we will provide you with a practical guide for decision-making. The best news first: the topic does not have to be a tale of woe - neither for manufacturers of medical devices nor for their suppliers.
Prologue
The company "Premier Components" has been around for many years. It has the facilities, the equipment, and the know-how to fill containers with liquids at its site. One of its customers is Medical Devices LLC, a manufacturer of medical products. A filling and packaging process has been carried out for them for five years. This process is almost fully automated on one of the company’s systems.Medical Devices LLC has now contacted Premier Components by telephone to ask whether process validation documents are available and have informed them that they need them as soon as possible. An employee, Vera Alid, took up this request and promised to take care of it. After several conversations with colleagues, Vera's head is spinning: does she even know what exactly she is looking for? Gradually, she begins to doubt this, as the statements from her colleagues are confusing and sometimes contradictory:
What exactly does the manufacturer mean when he wants to see process validation?
According to the GHTF Guidance Quality Management Systems - Process Validation Guidance, process validation is defined as follows: "establishing by objective evidence that a process consistently produces a result or product meeting its predetermined requirements." This definition has also been adopted by the US Food and Drug Administration (FDA) in 21 CFR Part 820.3. The company in our case study therefore wants evidence for thisstatement.Annex I (General Safety and Performance Requirements (GSPR)) of the Medical Device Regulation (EU) 2017/745 (MDR) states the following requirements:
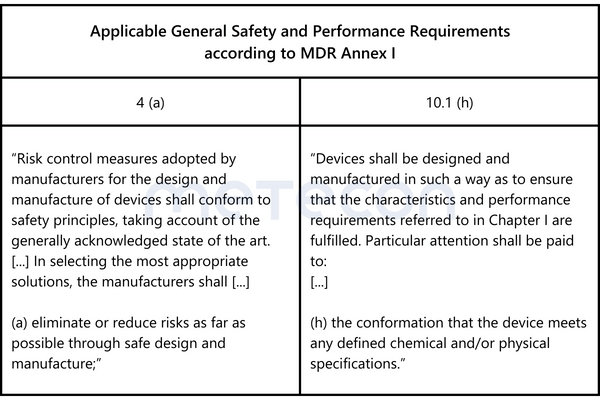
These formulations make it clear that risks during production must be minimized as far as possible. Without objective evidence that the manufacturing process consistently produces products with properties within the requirements, this requirement cannot be met.The GSPRs or the definitions of the GHTF Guidance /CFR 21 Part 820.3 do not contain specific indications of how this evidence should be presented.However, Chapter 5 "Conduct a Validation" of the GHTF Guidance contains recommendations/suggestions on this matter.Before proceeding with the actual implementation, the following question must be answered:
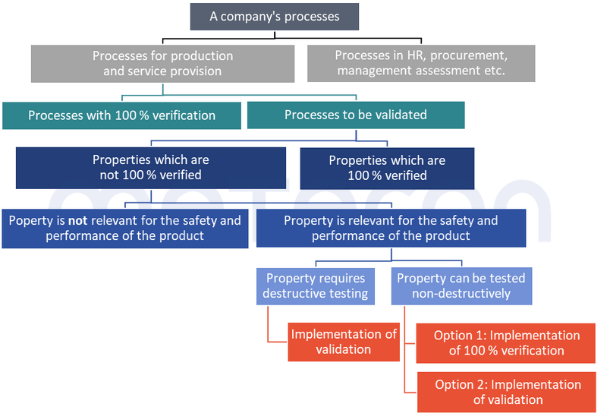
Which processes must be validated?
The GHTF Guideline specifies this, but something can also be read about it in ISO 13485:2016, 7.5.6 "Validation of processes for production and service provision". It stipulates that processes must be validated if the result cannot be verified by other measures before being put into use.A very similar requirement can be found in Chapter 6 Task 7 of the MDSAP Audit Approach and in CFR 21 Part 820.75 (a) .A process of which the result is 100 % verified, i.e. checked – for example whether each package contains instructions for use, does not need to be validated.But what about complex processes in which some results are not checked at all or only on a random basis, but which involve a 100 % check for other results?In such cases, validation would focus on those properties that are not or insufficiently checked (verified). However, the fact that these properties are 100 % verified should be justified in a comprehensible manner.
Validation or 100% verification?
Whether validation is carried out or a control measure for 100 % verification is built into the process is partly up to the company itself. For properties that require destructive testing, such as sterility, a 100 % verification is clearly not possible. However, for other properties, such as a fill quantity control, the company still has the option of making this decision. Economic reasons can play a role here. The costs of validation (personnel costs during validation, random sample testing, system availability) must be compared to the costs of implementing a 100 % verification.The relationships explained and the path to deciding whether a process needs to be validated are illustrated in the following diagram.
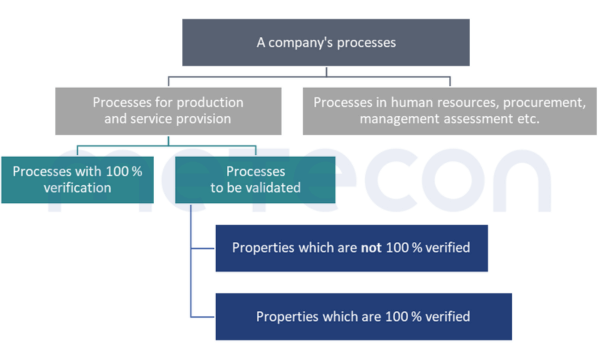
A word of caution: If only parts of the product properties are changed during processes, only the properties affected by the process need to be considered during validation. Nevertheless, it can of course happen that a property is unintentionally negatively altered by the process. Such aspects should be considered and documented in a risk analysis of the process. In this way, the procedure used in the validation can be justified on a risk-based basis.Example: two components are welded together. If this weld seam can have a safety-relevant impact on the product, the result of the welding must be considered in validation, for example with regard to strength. On the other hand,the geometric dimensions, of a hole far from the point where the weld was made, do not have to be measured.
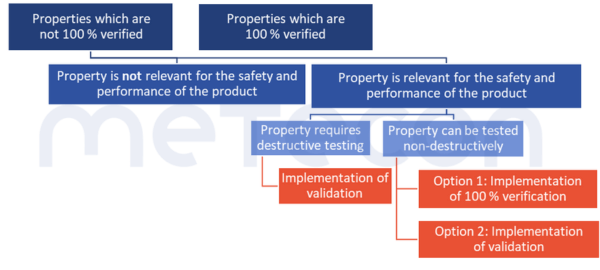
Are there exceptions?
1. No risk to safety and performance
There are properties that do not need to be verified to 100%, nor do they need to be considered within the scope of validation. This is the case when the absence or deviation from the specification poses no risk to the safety and performance of the product.The MDSAP Audit ApproachChapter 6 Task 7 states: "Processes that cannot be fully verified include processes where routine inspection and/or testing does not examine quality attributes essential to the proper functioning of the finished device."ISO 13485:2016 in 7.3.4 "Development Results" requires that such characteristics be defined.This could be the color of a housing, for example. If such characteristics exist, however, such a decision should be justified and documented.2. Duplicate effort only where absolutely necessary
There is also another option that may reduce or even completely eliminate the validation effort. Properties that are fully verified at one point in the overall process do not have to be considered at another point during validation. However, this only applies if the processes that follow the 100 % verification are highly unlikely to have a negative impact on the property that has been 100 % verified. Such decisions should also be fully documented to avoid difficulties in audits.Example: A packaging process is carried out by a machine. This prints the labels and sticks them onto the packaging. The system does not check whether the data on the labels is correct or whether a label is present on the product. The labeled product is packed manually by employees at a later stage. According to the work instructions, they must scan the label of each product and check whether the stored data (batch, production date) is correct. A missing or incorrect label would then be identified.
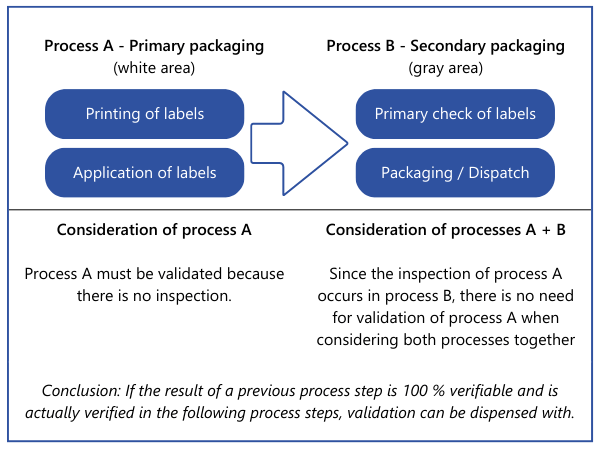
The procedure shown first lists all processes for production and service provision and explains where properties of the products are changed that are relevant to safety and performance. In principle, the order can also be reversed. All products and their safety and performance-relevant properties are listed. The processes involved in production and service provision are then described for each product. Ultimately, the same information is required.Please note: In this article, we talk about properties of "products". However, this does not only refer to end products. This is explicitly described in the MDSAP Audit approach Chapter 6 Task 7: "Process validation may apply to processes that generate components, subassemblies, or finished devices." Therefore, these processes must also be evaluated to determine whether validation needs to be carried out and, if so, for which one.
So how is the final decision made?
All this results in the following decision diagram for evaluating whether a process needs to be validated and, if so, which properties need to betaken into account:
Step 1: Documentation of results
In order to lay a foundation for the validation activities, an overview should be available of which processes are carried out in the company and which of these need to be validated. The term "Validation Master Plan" is widely used for such a document and is recommended by the GHTF Guidance and an FDA presentation. However, there is no regulatory requirement for such a document. If it does exist, it can also include responsibilities, timelines, and references to the documents created.And what happened next for V. Alid from KOMPONENTEN-KÖNIGE? Vera Alid also familiarized herself with the guidelines and regulations and gained a better understanding of how she should deal with the request from Medizinprodukte GmbH. In our next article, you will find out how we think she should proceed.
What you should definitely take away with you:
 Leon Weißenhorn
Leon Weißenhorn
Prologue
The company "Premier Components" has been around for many years. It has the facilities, the equipment, and the know-how to fill containers with liquids at its site. One of its customers is Medical Devices LLC, a manufacturer of medical products. A filling and packaging process has been carried out for them for five years. This process is almost fully automated on one of the company’s systems.Medical Devices LLC has now contacted Premier Components by telephone to ask whether process validation documents are available and have informed them that they need them as soon as possible. An employee, Vera Alid, took up this request and promised to take care of it. After several conversations with colleagues, Vera's head is spinning: does she even know what exactly she is looking for? Gradually, she begins to doubt this, as the statements from her colleagues are confusing and sometimes contradictory:
- "We don't need to do a process validation for the process - we perform a test for each batch at the end."
- "Process validations are only required for pharmaceutical companies."
- "The process has been running like this for a long time without any problems. Why do we have to submit documents now?"
- "What are we supposed to check during such a process validation? Everything? We don't have time for that!"
- "Can we introduce additional controls so that we don't have to validate the process?"
Validation in production – Part 1: Where to start
First of all, a very fundamental question must be answered:What exactly does the manufacturer mean when he wants to see process validation?
According to the GHTF Guidance Quality Management Systems - Process Validation Guidance, process validation is defined as follows: "establishing by objective evidence that a process consistently produces a result or product meeting its predetermined requirements." This definition has also been adopted by the US Food and Drug Administration (FDA) in 21 CFR Part 820.3. The company in our case study therefore wants evidence for thisstatement.Annex I (General Safety and Performance Requirements (GSPR)) of the Medical Device Regulation (EU) 2017/745 (MDR) states the following requirements:These formulations make it clear that risks during production must be minimized as far as possible. Without objective evidence that the manufacturing process consistently produces products with properties within the requirements, this requirement cannot be met.The GSPRs or the definitions of the GHTF Guidance /
Which processes must be validated?
The GHTF Guideline specifies this, but something can also be read about it in ISO 13485:2016, 7.5.6 "Validation of processes for production and service provision". It stipulates that processes must be validated if the result cannot be verified by other measures before being put into use.A very similar requirement can be found in Validation or 100% verification?
Whether validation is carried out or a control measure for 100 % verification is built into the process is partly up to the company itself. For properties that require destructive testing, such as sterility, a 100 % verification is clearly not possible. However, for other properties, such as a fill quantity control, the company still has the option of making this decision. Economic reasons can play a role here. The costs of validation (personnel costs during validation, random sample testing, system availability) must be compared to the costs of implementing a 100 % verification.The relationships explained and the path to deciding whether a process needs to be validated are illustrated in the following diagram.A word of caution: If only parts of the product properties are changed during processes, only the properties affected by the process need to be considered during validation. Nevertheless, it can of course happen that a property is unintentionally negatively altered by the process. Such aspects should be considered and documented in a risk analysis of the process. In this way, the procedure used in the validation can be justified on a risk-based basis.Example: two components are welded together. If this weld seam can have a safety-relevant impact on the product, the result of the welding must be considered in validation, for example with regard to strength. On the other hand,the geometric dimensions, of a hole far from the point where the weld was made, do not have to be measured.
Are there exceptions?
1. No risk to safety and performanceThere are properties that do not need to be verified to 100%, nor do they need to be considered within the scope of validation. This is the case when the absence or deviation from the specification poses no risk to the safety and performance of the product.The MDSAP Audit Approach
There is also another option that may reduce or even completely eliminate the validation effort. Properties that are fully verified at one point in the overall process do not have to be considered at another point during validation. However, this only applies if the processes that follow the 100 % verification are highly unlikely to have a negative impact on the property that has been 100 % verified. Such decisions should also be fully documented to avoid difficulties in audits.Example: A packaging process is carried out by a machine. This prints the labels and sticks them onto the packaging. The system does not check whether the data on the labels is correct or whether a label is present on the product. The labeled product is packed manually by employees at a later stage. According to the work instructions, they must scan the label of each product and check whether the stored data (batch, production date) is correct. A missing or incorrect label would then be identified.
The procedure shown first lists all processes for production and service provision and explains where properties of the products are changed that are relevant to safety and performance. In principle, the order can also be reversed. All products and their safety and performance-relevant properties are listed. The processes involved in production and service provision are then described for each product. Ultimately, the same information is required.Please note: In this article, we talk about properties of "products". However, this does not only refer to end products. This is explicitly described in the MDSAP Audit approach Chapter 6 Task 7: "Process validation may apply to processes that generate components, subassemblies, or finished devices." Therefore, these processes must also be evaluated to determine whether validation needs to be carried out and, if so, for which one.
So how is the final decision made?
All this results in the following decision diagram for evaluating whether a process needs to be validated and, if so, which properties need to betaken into account:Step 1: Documentation of results
In order to lay a foundation for the validation activities, an overview should be available of which processes are carried out in the company and which of these need to be validated. The term "Validation Master Plan" is widely used for such a document and is recommended by the GHTF Guidance and an FDA presentation. However, there is no regulatory requirement for such a document. If it does exist, it can also include responsibilities, timelines, and references to the documents created.And what happened next for V. Alid from KOMPONENTEN-KÖNIGE? Vera Alid also familiarized herself with the guidelines and regulations and gained a better understanding of how she should deal with the request from Medizinprodukte GmbH. In our next article, you will find out how we think she should proceed.What you should definitely take away with you:
- The properties relevant to the safety and performance of the product must be known.
- Knowledge about the processes and controls carried out is essential.
- The entire process chain for the product should be considered. This way, properties can be identified that are 100% verified elsewhere ´´.
- Instead of validation, measures for 100% verification can sometimes be implemented.
- Document your decisions including a justification.
Our blog posts are researched and created with the utmost care, but are only snapshots of the regulations, which are constantly changing. We do not guarantee that older content is still current or meaningful. If you are not sure whether the article you have read on this page still corresponds to the current state of regulation, please contact us: we will quickly place your topic in the current context.
Regulatory Affairs & Technical Documentation



