
The Equivalence of Medical Devices Under MDR: A guide
23/04/2024
Do you have any questions about the article or would you like to find out more about our services? We look forward to hearing from you!Make a non-binding enquiry now
Demonstrating equivalence of medical devices in accordance with the requirements of the Medical Device Regulation (EU) 2017/745 (MDR) is a critical aspect that can pose significant challenges for manufacturers. Especially within the context of clinical evaluation under the MDR, it becomes apparent how crucial it is to adequately demonstrate equivalence. Notified Bodies emphasize the importance of this process, which not only contributes to ensuring conformity but also leads to time and cost savings.But how exactly is equivalence demonstrated according to the MDR? And what hurdles and pitfalls need to be considered? The following article provides a comprehensive overview of the requirements, processes, and implications of demonstrating equivalence under the MDR.Notified Bodies identify confusion surrounding equivalent products as a stumbling block on the path to MDR compliance, which causes many manufacturers to fail. Under the MDD, equivalence was demonstrated according to Annex A1 of MEDDEV 2.7.1 rev. 4. The MDR tightens the requirements in Annex XIV, and the Medical Device Coordination Group contributes further clarification in their guidance document MDCG 2020-5.Why demonstrate equivalence
For many medical devices, clinical data are required to demonstrate conformity with the relevant essential safety and performance requirements as outlined in Annex I of the MDR. If the clinical evaluation lacks sufficient clinical data, these may need to be generated from the use of the medical device within its intended purpose.Clinical trials can be time- and cost-intensive and require additional expertise that may not be available within the company, such as in design, defining endpoints, ensuring data quality, and evaluating results.One possible solution is establishing equivalence. If this is proven, clinical data from the equivalent product may be used for the clinical evaluation according to Annex XIV, Section 3 of the MDR.However, this demonstration is not always straightforward.Please note: Establishing equivalence only enables the transfer of clinical data. Non-clinical data, such as biocompatibility, mechanical and functional tests, benchmark tests, and animal studies, are not included here.
How to demonstrate equivalence?
For the clinical evaluation of implantable products and Class III devices, the biggest hurdle is the requirement in Article 61(5) for contractually regulated unrestricted access to the technical documentation of the medical device used to demonstrate equivalence.Since competitors are rarely willing to engage in such information exchange, this option remains unavailable to manufacturers of most implantable medical devices and/or Class III medical devices. An exception exists if, as described in Article 61(4), equivalence with a product from the same manufacturer is to be demonstrated.For demonstrating equivalence with products other than Class III or implantable medical devices, no contract between manufacturers is required. However, access to information about the equivalent product should be verified. The sources should be included in the clinical evaluation.Equivalence is demonstrated according to Annex XIV, Section 3 of the MDR. The requirements of Annex A1 of MEDDEV 2.7.1 rev. 4 are not applicable under the MDR. The characteristics to be compared are summarized in the following overview table.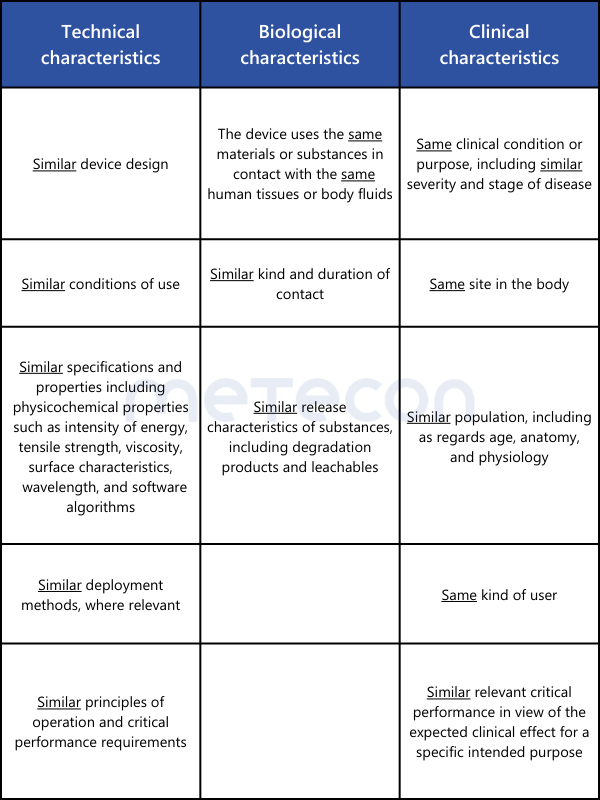
What to consider?
As can be seen from the overview table, some points must indeed be "identical" for the equivalence of two medical devices to be correctly demonstrated. If a difference is found here, equivalence is not established. In the remaining points, where only similarity is required, all differences must be individually evaluated. It must be scientifically justified whether these differences have significant impacts on the clinical performance or safety of the medical devices.For demonstrating the equivalence of two medical devices without a medical purpose (examples from Annex XVI of the MDR: contact lenses, devices for liposuction, lipolysis, or lipoplasty), the explanations from MDCG 2023-6 can be utilized.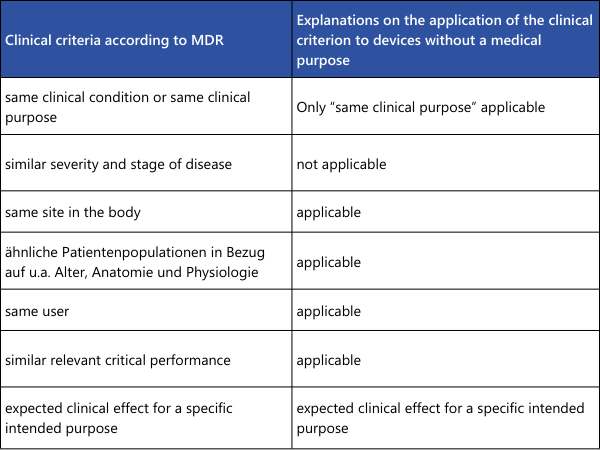
Similar medical devices
A common mistake in practice is using clinical data from a similar medical device without demonstrating equivalence. This leads to a deviation in conformity assessment. A clear distinction must be made here. MDCG 2020-6 refers to similar products as products belonging to a generic group and cites the definition from Article 2(7) of the MDR, which defines a "generic group of products" as a group of products with the same or similar intended purposes or with technological similarities that can be classified generally, i.e., without considering specific features.Based on clinical data generated with such products, acceptance criteria for the clinical performance or safety of generic medical devices can be presented according to the state of the art. Information from literature on design development, safety, and performance profile can help assess whether it is a well-established technology.For medical devices based on a well-established technology, clinical data from similar products can be supportive for demonstrating conformity with the relevant essential safety and performance requirements. However, "supportive" means that the evidence of a performance or safety aspect must not solely rely on the data from similar products.Predicate devices
By the way: The MDR is not the only set of medical device regulation to define "equivalence", and other jurisdictions have also used this concept, even if it is used and defined differently. An example is the "substantial equivalence" comparison with the "predicate device," which is the cornerstone of the FDA's 510(k). Here it is important to understand how "equivalence" is dewfined and used in each jurisdiction to ensure you are compliant.What comes next?
Equivalent or similar products, whose clinical data have been used for the conformity assessment of a product, must continue to be monitored within the framework of Post-Market Clinical Follow-up (PMCF) measures. This is reflected in the templates proposed by the Medical Device Coordination Group (MDCG) for the PMCF plan (MDCG 2020-7) and the PMCF report (MDCG 2020-8). This aims to ensure continuous updating of the benefit-risk ratio.Do you have questions about the equivalence of medical devices, clinical evaluation or other aspects relating to the clinical use of your medical device? Please feel free to contact me. Together with my colleagues from Clinical Affairs, I will be happy to support you in all matters.
Why demonstrate equivalence
For many medical devices, clinical data are required to demonstrate conformity with the relevant essential safety and performance requirements as outlined in Annex I of the MDR. If the clinical evaluation lacks sufficient clinical data, these may need to be generated from the use of the medical device within its intended purpose.Clinical trials can be time- and cost-intensive and require additional expertise that may not be available within the company, such as in design, defining endpoints, ensuring data quality, and evaluating results.One possible solution is establishing equivalence. If this is proven, clinical data from the equivalent product may be used for the clinical evaluation according to Annex XIV, Section 3 of the MDR.However, this demonstration is not always straightforward.Please note: Establishing equivalence only enables the transfer of clinical data. Non-clinical data, such as biocompatibility, mechanical and functional tests, benchmark tests, and animal studies, are not included here.How to demonstrate equivalence?
For the clinical evaluation of implantable products and Class III devices, the biggest hurdle is the requirement in Article 61(5) for contractually regulated unrestricted access to the technical documentation of the medical device used to demonstrate equivalence.Since competitors are rarely willing to engage in such information exchange, this option remains unavailable to manufacturers of most implantable medical devices and/or Class III medical devices. An exception exists if, as described in Article 61(4), equivalence with a product from the same manufacturer is to be demonstrated.For demonstrating equivalence with products other than Class III or implantable medical devices, no contract between manufacturers is required. However, access to information about the equivalent product should be verified. The sources should be included in the clinical evaluation.Equivalence is demonstrated according to Annex XIV, Section 3 of the MDR. The requirements of Annex A1 of MEDDEV 2.7.1 rev. 4 are not applicable under the MDR. The characteristics to be compared are summarized in the following overview table.What to consider?
As can be seen from the overview table, some points must indeed be "identical" for the equivalence of two medical devices to be correctly demonstrated. If a difference is found here, equivalence is not established. In the remaining points, where only similarity is required, all differences must be individually evaluated. It must be scientifically justified whether these differences have significant impacts on the clinical performance or safety of the medical devices.For demonstrating the equivalence of two medical devices without a medical purpose (examples from Annex XVI of the MDR: contact lenses, devices for liposuction, lipolysis, or lipoplasty), the explanations from MDCG 2023-6 can be utilized.What won’t work?
Products with different intended purposes
Equivalence cannot be demonstrated between a product without a medical purpose and a comparable product with a medical purpose. If a product/device without a medical purpose is compared to a product that has both medical and non-medical purposes, only the features related to the non-medical purpose may be considered. Consequently, even after establishing equivalence, only the clinical data associated with the non-medical use may be used.Similar medical devices
A common mistake in practice is using clinical data from a similar medical device without demonstrating equivalence. This leads to a deviation in conformity assessment. A clear distinction must be made here. MDCG 2020-6 refers to similar products as products belonging to a generic group and cites the definition from Article 2(7) of the MDR, which defines a "generic group of products" as a group of products with the same or similar intended purposes or with technological similarities that can be classified generally, i.e., without considering specific features.Based on clinical data generated with such products, acceptance criteria for the clinical performance or safety of generic medical devices can be presented according to the state of the art. Information from literature on design development, safety, and performance profile can help assess whether it is a well-established technology.For medical devices based on a well-established technology, clinical data from similar products can be supportive for demonstrating conformity with the relevant essential safety and performance requirements. However, "supportive" means that the evidence of a performance or safety aspect must not solely rely on the data from similar products.Predicate devices
By the way: The MDR is not the only set of medical device regulation to define "equivalence", and other jurisdictions have also used this concept, even if it is used and defined differently. An example is the "substantial equivalence" comparison with the "predicate device," which is the cornerstone of the FDA's 510(k). Here it is important to understand how "equivalence" is dewfined and used in each jurisdiction to ensure you are compliant.What comes next?
Equivalent or similar products, whose clinical data have been used for the conformity assessment of a product, must continue to be monitored within the framework of Post-Market Clinical Follow-up (PMCF) measures. This is reflected in the templates proposed by the Medical Device Coordination Group (MDCG) for the PMCF plan (MDCG 2020-7) and the PMCF report (MDCG 2020-8). This aims to ensure continuous updating of the benefit-risk ratio.Do you have questions about the equivalence of medical devices, clinical evaluation or other aspects relating to the clinical use of your medical device? Please feel free to contact me. Together with my colleagues from Clinical Affairs, I will be happy to support you in all matters.
Our blog posts are researched and created with the utmost care, but are only snapshots of the regulations, which are constantly changing. We do not guarantee that older content is still current or meaningful. If you are not sure whether the article you have read on this page still corresponds to the current state of regulation, please contact us: we will quickly place your topic in the current context.




