
Switzerland: New regulation of in vitro diagnostics by IvDO
07/02/2023
Do you have any questions about the article or would you like to find out more about our services? We look forward to hearing from you!Make a non-binding enquiry now
In the past, the Mutual Recognition Agreement (MRA) between Switzerland and the EU has facilitated cross-border trade in medical devices/in vitro diagnostic devices. When the EU In Vitro Diagnostic Regulation 2017/746 (IVDR) came into force in May 2022, a new agreement became necessary. However, the agreement on mutual recognition of conformity assessments was no longer updated.Switzerland has since become an EU third country, and its market is no longer freely accessible. Barrier-free mutual market access and common market surveillance are no longer guaranteed. To place IVDs on the market, EU manufacturers need a Swiss authorized representative and importer. This requirement arises from the Swiss Medical Devices Ordinance (MedDO; SR 812.213).Medical devices that are placed on the market or put into service in Switzerland must meet the requirements of the MedDO or the Ordinance on In-vitro Diagnostic Medical Devices (IvDO, SR 812.219 ). In this article, you will learn more about the regulation of in-vitro diagnostic devices in the Swiss market.
Additional info: Clinical trials with in vitro diagnostic devices will be regulated in the ClinO-MD as of May 26, 2022, and no longer in the Ordinance on Clinical Trials (ClinO). The adjustments to the ClinO-MD enable comprehensive regulation of research on humans with all medical devices, including IVDs. This will tighten the requirements for research regarding safety and performance demonstrations for in vitro diagnostic devices in line with European regulations.
Source: https://www.swiss-medtech.ch/en/news/overview-ivdo-dates-and-deadlines
Importers and distributors established in Switzerland must submit the following form to Swissmedic:
EU/EEA based manufacturers of IVDs do not have to notify their products separately to Swissmedic if they are placed on the market in Switzerland. Provided that the products are IVDR compliant (CE) and a person based in Switzerland has been registered as CH-REP!As of May 27, 2022, no IVDs will be newly registered on the Swiss market according to IVDD (98/79/EC). IVDs still on the market (legacy devices) must not undergo any significant changes!
Do not lose sight of the transition deadlines. The deadlines for designating a CH-REP expire already in March for Class C & D products and July for Class A products.You can also find more detailed information on the page Swissmedic.ch in the factsheet "FAQ – Notification of in-vitro diagnostic medical devices".Do you need assistance in preparing and submitting the required documents to Swissmedic? Do you appreciate support in finding a CH-REP? We will be happy to assist you and provide you with low-barrier access to the Swiss market. We are also your contact for further questions regarding the approval of your IVDs. We look forward to hearing from you!
Best Regards
 Dunja Schildge-Reichman
Dunja Schildge-Reichman
The new IvDO applies since May 26, 2022
On May 4, 2022, the Swiss Federal Council adopted the new IvDO and the amending decree of the Ordinance on Clinical Trials with Medical Devices (ClinO-MD). They are the final stage of the adaptation of Swiss medical device law and pursue several goals:- Improve patient safety through more stringent conformity assessment requirements,
- post-market surveillance and
- alignment with the new European Union regulations.
Additional info: Clinical trials with in vitro diagnostic devices will be regulated in the ClinO-MD as of May 26, 2022, and no longer in the Ordinance on Clinical Trials (ClinO). The adjustments to the ClinO-MD enable comprehensive regulation of research on humans with all medical devices, including IVDs. This will tighten the requirements for research regarding safety and performance demonstrations for in vitro diagnostic devices in line with European regulations.
The situation in the EU
The implementation of IVDR could not take place by the effective date of May 26, 2022, due to the challenges of the Covid-19 pandemic, the limited capacity of notified bodies, and the complexity of the regulation itself. Therefore, on January 25, 2022, the EU issued "Regulation (EU) 2022/112 amending Regulation (EU) 2017/746 as regards transitional provisions for certain in vitro diagnostic medical devices and the deferred application of conditions for in-house devices”. With these transitional periods, the EU intends to prevent an impending supply gap. They are graded according to risk classes and last until 2027 at the longest.Transitional provisions in Switzerland
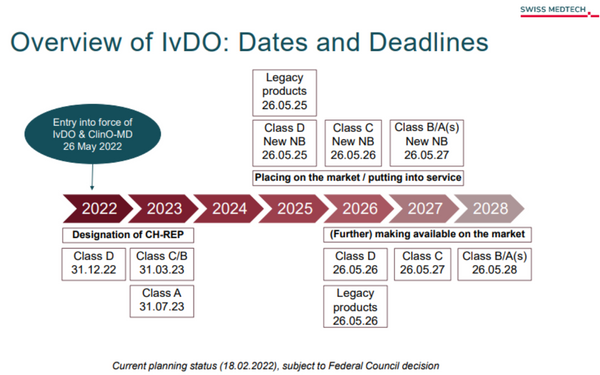
These EU’s new transition periods are also considered accordingly in the IvDO. The IvDO additionally provides for various transition periods and measures to ensure the continued supply of safe IVDs from the EU to Switzerland. The following measures are planned:1. EU certificates of conformity are recognized!2. Registration of economic operators (manufacturers, importers and authorized representatives): The registration is carried out by Swissmedic. There, they receive a unique registration number, the Swiss Single Registration Number - CHRN.3. The notification of serious incidents and safety reports is also reported to Swissmedic. This notification is made by the designated CH-REP for the EU-based manufacturers of IVDs.4. The establishment of a Swiss authorized representative (CH-REP) for foreign manufacturers should help Swissmedic to maintain market surveillance despite exclusion from the network of EU authorities. Longer transition periods apply for this (see figure).5. Facilitation of the labeling obligation: for all products that are handled by professionals, the IvDO allows for an alternative CH-REP indication on a document accompanying the IVD (e. g. a delivery note). This does not apply to products intended for self-testing that are placed on the market in accordance with the new law. Deadline extension until March 31, 2025.Figure transition periods: Overview of the IvDO: dates and deadlines:Source: https://www.swiss-medtech.ch/en/news/overview-ivdo-dates-and-deadlines
Authorization/notification of in vitro diagnostics according to IvDO in Switzerland
Swiss-based manufacturers of IVDs must inform Swissmedic as soon as they wish to initially place their products on the Swiss market.I. IVDs of classes D, C, B and A sterile must be reported individually. Swissmedic provides two forms for this purpose:- Form for notification of manufacturers according to Art. 90 para.1 IvDO,
- Form for certification data of in-vitro diagnostics Art. 90 para. 1 IvDO (IVDR Annex IX-XI).
- the certificate on the conformity assessment procedures carried out (EC certificates), and
- the instructions for use, as well as for products for self-testing and near-patient testing (POCT) – additionally the layout of the outer packaging.
- Form for the notification of manufacturers according to Art. 90 par.1 IvDO
Importers and distributors established in Switzerland must submit the following form to Swissmedic:
- Form notification of repackaged/relabeled in vitro diagnostic medical devices according to Art. 46 para. 4 and Art. 47 para. 4 IvDO.
- Medical analytical test procedures developed in-house and performed with own (not CE-marked) reagents.
- Medical analytical test procedures based on standard procedures or published procedures, performed with own (not CE-marked) reagents.
- Acquired but not intended for medical use test procedures (e.g., "Research Use Only/RUO" procedures) that have been (further) developed by the healthcare facility for medical analytical use.
- IVD instruments manufactured in-house.
- IVD software developed in-house.
EU/EEA based manufacturers of IVDs do not have to notify their products separately to Swissmedic if they are placed on the market in Switzerland. Provided that the products are IVDR compliant (CE) and a person based in Switzerland has been registered as CH-REP!As of May 27, 2022, no IVDs will be newly registered on the Swiss market according to IVDD (98/79/EC). IVDs still on the market (legacy devices) must not undergo any significant changes!
Conclusion
Use the remaining time to get to grips with the new regulations and make sure you take all the necessary steps to achieve compliance in good time.Do not lose sight of the transition deadlines. The deadlines for designating a CH-REP expire already in March for Class C & D products and July for Class A products.You can also find more detailed information on the page Swissmedic.ch in the factsheet "FAQ – Notification of in-vitro diagnostic medical devices".Do you need assistance in preparing and submitting the required documents to Swissmedic? Do you appreciate support in finding a CH-REP? We will be happy to assist you and provide you with low-barrier access to the Swiss market. We are also your contact for further questions regarding the approval of your IVDs. We look forward to hearing from you!
Best Regards
Our blog posts are researched and created with the utmost care, but are only snapshots of the regulations, which are constantly changing. We do not guarantee that older content is still current or meaningful. If you are not sure whether the article you have read on this page still corresponds to the current state of regulation, please contact us: we will quickly place your topic in the current context.
Regulatory Affairs Expert



