
Entscheidungshilfe: Brauchen Sie eine klinische Leistungsstudie nach IVDR?
23/07/2021
Do you have any questions about the article or would you like to find out more about our services? We look forward to hearing from you!Make a non-binding enquiry now
From our archives (23/07/2021)
Die neue IVD-Verordnung (EU) 2017/746 (IVDR) stellt weitaus höhere Anforderungen an die Leistungsbewertung von IVDs als die IVD-Richtlinie 98/79/EG, die am 26.5.2022 durch die IVDR abgelöst wird. Gerade im Bereich der klinischen Leistungsbewertung herrscht große Unsicherheit, wie bereits vorhandene klinische Daten in die neue klinische Leistungsbewertung eingebunden werden können und wann eine neue klinische Leistungsstudie nach Vorgaben der IVDR benötigt wird. Dr. Sandra Reuter und Dr. Caroline Arnold helfen Ihnen, den Überblick zu behalten.Die Leistungsbewertung nach IVDR ist auf drei Säulen aufgebaut: dem Nachweis der wissenschaftlichen Validität, der Analyseleistung und der klinischen Leistung. Durch die Bewertung dieser drei Säulen kann der klinische Nachweis erbracht werden, d. h. es kann beurteilt werden, ob das Produkt sicher ist und den angestrebten klinischen Nutzen bei bestimmungsgemäßer Verwendung erzielt.Definition "klinische Leistung"
Die IVDR definiert die klinische Leistung als "die Fähigkeit des Produktes Ergebnisse zu liefern, die mit einem bestimmten klinischen Zustand oder physiologischen oder pathologischen Vorgang oder Zustand bei einer bestimmten Zielbevölkerung und bestimmten vorgesehenen Anwendern korrelieren". Die nach Anhang I Abschnitt 9.1 b) geforderten Parameter zur Bestimmung der klinischen Leistung sind:
- diagnostische Sensitivität,
- diagnostische Spezifität,
- positiver prädiktiver Wert,
- negativer prädiktiver Wert,
- Likelihood-Verhältnis sowie
- erwartete Werte bei nicht betroffenen und betroffenen Bevölkerungsgruppen.
Wenn einer oder mehrere dieser Parameter für das Produkt nicht anwendbar sind, muss dies unbedingt begründet werden! Der Nachweis der klinischen Leistung eines Produkts beruht auf einer der folgenden Quellen oder einer Kombination dieser Quellen:
- klinische Leistungsstudien,
- wissenschaftliche Literatur, die einem Peer-Review unterzogen wurde,
- aus diagnostischen Routinetests gewonnene Erfahrungen, die veröffentlicht wurden.
Weiterhin fordert die IVDR, dass auf die Durchführung klinischer Leistungsstudien gemäß Anhang XIII Teil A Abschnitt 2 nur dann verzichtet werden kann, wenn es ausreichende Gründe dafür gibt, auf andere Quellen klinischer Leistungsdaten zurückzugreifen.
Wann Sie KEINE klinische Leistungsstudie nach IVDR brauchen
Was sind ausreichende Gründe, um auf eine klinische Leistungsstudie gemäß Anhang XIII verzichten zu können? Einen wichtigen Hinweis hierzu gibt die GHTF (Global Harmonization Task Force), die den Bedarf einer klinischen Leistungsstudie vom Standardisierungsgrad des jeweiligen Tests abhängig macht (GHTF/SG5/N7:2012: Clinical Evidence for IVD medical devices – Scientific Validity Determination and Performance Evaluation). Demnach ist eine klinische Leistungsstudie für bereits etablierte und standardisierte Tests nicht erforderlich, um die Konformität mit den relevanten Grundlegenden Sicherheits- und Leistungsanforderungen nachzuweisen. Für etablierte, aber nicht standardisierte Produkte ist sie häufig erforderlich und für neuartige Teste höchstwahrscheinlich erforderlich.Weiterhin gibt es IVDs, auf die klinische Leistungsstudien aufgrund ihrer Zweckbestimmung nicht anwendbar sind, z. B. wenn das Produkt/Test-Kit nicht zur Diagnose einer Erkrankung, sondern lediglich zum Monitoring eines Markers genutzt wird.Sollte es sich bei Ihrem Produkt um ein äquivalentes Produkt zu einem auf dem Markt befindlichen Produkt handeln, so können Sie dies als Vergleich heranziehen und müssen keine eigene klinische Leistungsstudie initiieren. Damit Sie den Vergleich auf technischer, biologischer und klinischer Ebene adäquat durchführen können, benötigen Sie allerdings Einsicht in alle relevanten Daten aus der technischen Dokumentation dieses Vergleichsprodukts. Dies wird in den meisten Fällen schlicht nicht möglich sein, es sei denn, es handelt sich um ein von Ihnen hergestelltes Produkt. Gezielte Anforderungen zur Demonstration der Äquivalenz, wie wir sie in der Medizinprodukteverordnung (EU) 2017/745 in Anhang XIV Abschnitt 3 finden, fehlen in der IVDR allerdings. Somit ist dieser Weg nur für sehr wenige Produkte gangbar. Auch wenn Sie begründen können, warum für Ihr Produkt keine klinische Leistungsstudie erforderlich ist, müssen Sie dennoch die klinische Leistung durch eine Recherche wissenschaftlicher Literatur belegen, indem Sie zu Ihrem Produkt publizierte Daten analysieren und bewerten. Dies können klinische Daten sein oder auch andere publizierte Daten wie z. B. Ringversuche. Falls Sie Ihr Produkt bereits unter der IVD-Richtlinie 98/79/EG in Verkehr gebracht haben, haben Sie wahrscheinlich eigene Studiendaten zur Ermittlung der klinischen Parameter wie diagnostische Sensitivität und Spezifität vorliegen, die bislang als Nachweis der klinischen Leistung Ihres Produktes dienten. Diese Daten können Sie, soweit diese immer noch aktuell sind, auch unter der IVDR benutzen, allerdings dürfen Sie diese Studien nicht als „klinische Leistungsstudien“ im Sinne der IVDR deklarieren, da sie sehr wahrscheinlich nicht die in Anhang XIII Teil A Abschnitt 2 beschriebenen komplexen Anforderungen erfüllen. Sie können diese Daten aber weiterhin unter „Sonstige Leistungsstudien“ gemäß Anhang XIII Teil A Abschnitt 3 führen und damit Ihre Argumentation stärken, warum Sie keine neue klinische Leistungsstudie durchführen müssen.Alle Daten und Erkenntnisse zur klinischen Leistung fassen Sie im „Bericht über die klinische Leistung“ zusammen. Achten Sie auf schlüssige Begründungen, wenn Sie einzelne Bausteine der klinischen Leistung nicht für anwendbar halten, und vergessen Sie nicht die Schlussfolgerungen aus den von Ihnen erhobenen bzw. bewerteten Daten.
Wann Sie eine klinische Leistungsstudie brauchen
Wenn Sie die klinische Leistung Ihres Produkts nicht anhand der wissenschaftlichen Literatur oder publizierter Routineteste oder anderer Quellen demonstrieren können, dann müssen Sie eine klinische Leistungsstudie nach Anhang XIII bzw. Anhang XIV (interventionelle klinische Leistungsstudien) durchführen. Für neuartige Marker oder neuartige Technologien ist demnach eine Leistungsstudie immer erforderlich. Bisher sind die regulatorischen Anforderungen an klinische Leistungsstudien sowie Genehmigungsverfahren für In-vitro-Diagnostika im Medizinproduktegesetz (MPG) und der Verordnung über klinische Prüfungen von Medizinprodukten (MPKPV) geregelt. Zusätzlich sind die Anforderungen an Planung, Durchführung, Auswertung und Dokumentation von Leistungsstudien in der DIN EN 13612:2002-08 festgehalten. In Zukunft sind alle relevanten Anforderungen in der IVDR zu finden und die DIN EN 13612:2002-08 wird ergänzt durch die ISO 20916:2019-05, die viel mit der DIN EN 14155:2003-11 für Medizinprodukte gemein hat. Der Vorteil besteht darin, sich die Erfahrung aus der Medizinproduktewelt bezüglich Studienplanung, Durchführung und Dokumentation zunutze zu machen und das Rad nicht neu erfinden zu müssen. Im aktuellen Corrigendum zur IVDR wird die Umsetzung der ISO 20916 in Erwägungsgrund 66 ebenfalls bereits gefordert. Zu beachten sind dabei die Genehmigungen bei Bundesoberbehörde und Ethikkommission, die je nach Studien- und Probenart und den damit verbundenen Risiken für den Patienten einzuholen sind. Danach richtet sich auch der Umfang der Studienplanung und Dokumentation, die in Artikel 56 - 76 sowie in Anhang XIII und XIV der IVDR beschrieben sind. Unser Tipp: Nutzen Sie nachfolgend unser Schaubild als Entscheidungshilfe, das Sie zu den entsprechenden Anforderungen der IVDR führt. Schaubild: Übersicht der IVDR-Anforderungen an klinische Leistungsstudien
Für größere Ansicht bitte Grafik anklicken
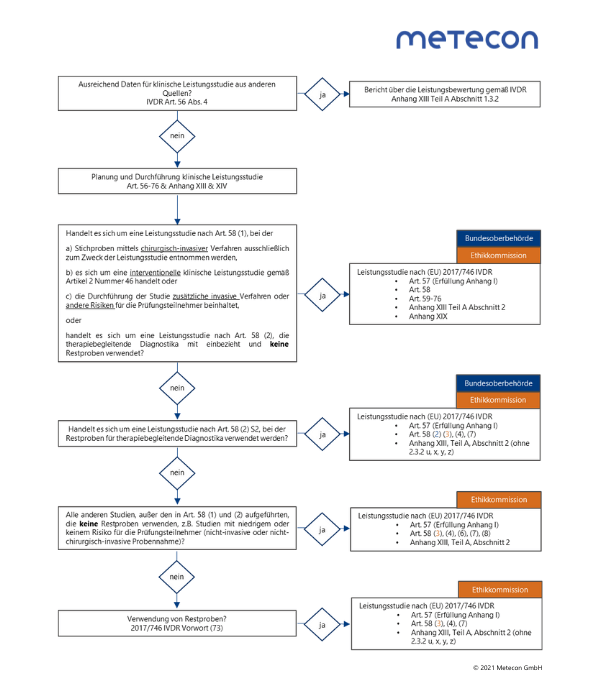 © 2021/07 Metecon GmbHSie haben Fragen oder benötigen Unterstützung bei der Planung Ihrer klinischen Leistungsstudie oder bei der Erstellung der von der IVDR geforderten Dokumente zur Leistungsbewertung? Wir freuen uns, von Ihnen zu hören!Herzliche Grüße
© 2021/07 Metecon GmbHSie haben Fragen oder benötigen Unterstützung bei der Planung Ihrer klinischen Leistungsstudie oder bei der Erstellung der von der IVDR geforderten Dokumente zur Leistungsbewertung? Wir freuen uns, von Ihnen zu hören!Herzliche Grüße
Our blog posts are researched and created with the utmost care, but are only snapshots of the regulations, which are constantly changing. We do not guarantee that older content is still current or meaningful. If you are not sure whether the article you have read on this page still corresponds to the current state of regulation, please contact us: we will quickly place your topic in the current context.

Team Lead IVD
Regulatory Affairs & Technical Documentation




