
Customized Medicine - Custom-made devices under the Medical Device Regulation (MDR)
29/01/2024
Do you have any questions about the article or would you like to find out more about our services? We look forward to hearing from you!Make a non-binding enquiry now
Medical devices under the Medical Device Regulation (EU) 2017/745 (MDR) encompass a wide range of products, from disposable syringes produced in the millions to magnetic resonance imaging devices, which are significantly less common with approximately 2900 devices in Germany (as of 2021). However, there are also products that are truly unique and produced only once: custom-made devices.But what exactly constitutes a custom-made device, and what needs to be considered? In the following, we describe what defines custom-made devices according to MDR and the implications for manufacturers whose products fall under this definition.
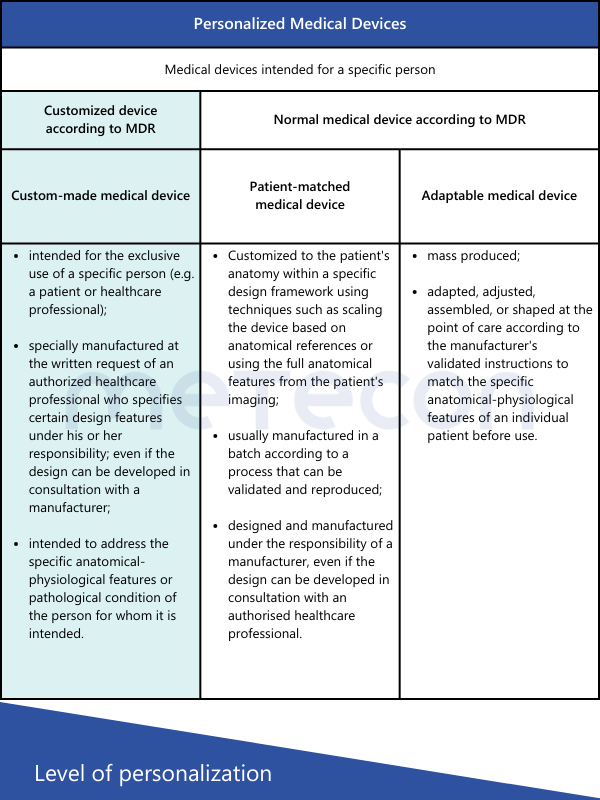
From these definitions, the following conclusions can be drawn regarding customized devices:
The MDR only distinguishes between "normal" medical devices and custom-made devices, therefore there are only special requirements for these in the MDR. While the MDCG and IMDRF provide additional definitions, these do not have regulatory implications for individual requirements.
Best RegardsLeon Weißenhorn
 Leon Weißenhorn
Leon Weißenhorn
What are custom-made devices?
MDR defines custom-made devices as follows in Article 2, Point 3:"custom-made device" means any device specifically made in accordance with a written prescription of any person authorised by national law by virtue of that person's professional qualifications which gives, under that person's responsibility, specific design characteristics, and is intended for the sole use of a particular patient exclusively to meet their individual conditions and needs.However, mass-produced devices which need to be adapted to meet the specific requirements of any professional user and devices which are mass-produced by means of industrial manufacturing processes in accordance with the written prescriptions of any authorised person shall not be considered to be custom-made devices;This definition explicitly states which types of devices are not custom-made. The International Medical Device Regulators Forum (IMDRF) has established its own definitions, also adopted by the MDCG 2021-3. In addition to the term "custom-made," other terms such as "personalized medical device," "patient-matched medical device," and "adaptable medical device" are mentioned. For those encountering such medical devices for the first time, the question arises: Are these terms synonymous, or what are the differences?The table below provides a brief comparison of the various definitions according to IMDRF and their characteristics.From these definitions, the following conclusions can be drawn regarding customized devices:
- All devices produced in series, even if they meet individual user requirements, are not considered custom-made.
- The device must be requested based on a written prescription from an authorized person to be considered custom-made.
- This person must specify the specific design of the device (e.g., number and position of screws, geometric dimensions) and is also responsible for it.
- The device must be intended for a specific patient to be considered custom-made.
The MDR only distinguishes between "normal" medical devices and custom-made devices, therefore there are only special requirements for these in the MDR. While the MDCG and IMDRF provide additional definitions, these do not have regulatory implications for individual requirements.
What should manufacturers of custom-made devices pay attention to?
For manufacturers of custom-made devices, some otherwise necessary activities are eliminated, some change, and others are added. Some of these activities are explained in more detail below.Note: Special considerations apply to Class III or implantable custom-made devices, which are not addressed in this article.Aspects that do not change (not a complete list)
- The manufacturer is still obligated to comply with basic safety and performance requirements (MDCG 2021-3, Point 8).
- A conformity assessment procedure is conducted (MDCG 2021-3, Point 9).
- Post-Market Surveillance (PMS), Post-Market Clinical Follow-up (PMCF ), risk management, and clinical evaluation must be conducted (MDCG 2021-3, Point 8).
- A Person Responsible for Regulatory Compliance (PRRC) is required (MDR: Article 15).
Aspects that are considered differently (not a complete list)
- No EU Declaration of Conformity is issued. Therefore, no CE marking is affixed to the product. The necessary conformity assessment can only be performed through the procedure outlined in Annex XIII (MDR: Article 10, Point 6, Article 20, Point 1, Article 21, Point 1).
- Collaboration with a notified body in the context of the conformity assessment is not necessary for Class IIa and IIb (MDCG 2021-3, Point 9).
- No Unique Device Identification (UDI) must be assigned or affixed (MDR: Recitals point 42, Article 27 point 1, 3, Article 29 point 1,2,4).
- The competent authorities may require the manufacturer to provide them with a list containing all custom-made devices that have been placed on the market in the respective territory (MDR: Article 21, point 2).
- The manufacturer, authorized representative, and importer do not have to register in the electronic system referred to in Article 30 (EUDAMED) (MDR: Article 30, point 3; Article 31, point 1).
- The PRRC also does not need to register in EUDAMED (MDCG 2021-3, Point 9).
- In contrast to the usual practice, it is possible in the case of custom-made devices to prove the PRRC's qualification through two years of professional experience in a relevant area of manufacturing (MDR: Article 15, point 1).
- The product and packaging must be labelled "custom-made device" (MDR: Annex I 23.2 and 23.3).
- Custom-made devices must be accompanied by a declaration according to Annex XIII, Section 1 of MDR (MDR: Article 21, Point 2). The associated user or patient must be identifiable by name, acronym, or numerical code (MDR: Article 21, Point 2).
- No technical documentation is created. However, documentation according to Annex XIII, Section 2 is required (MDR: Article 10, Point 5).
Conclusion
As with most other regulatory considerations, when dealing with custom-made devices under the MDR, many details must be considered. The classification of a medical device as a custom-made device brings about some changes for manufacturers – neither unmanageable obstacles nor irrelevant side issues.Do you need assistance with the conformity assessment procedure for your custom-made device? Or are you currently dealing with another complex issue related to Clinical Affairs or Technical Documentation and are in search of experienced professionals? Please feel free to contact us anytime. Together, we will find the best solution for your specific needs.Best RegardsLeon Weißenhorn
Our blog posts are researched and created with the utmost care, but are only snapshots of the regulations, which are constantly changing. We do not guarantee that older content is still current or meaningful. If you are not sure whether the article you have read on this page still corresponds to the current state of regulation, please contact us: we will quickly place your topic in the current context.
Regulatory Affairs & Technical Documentation



