
Digitale Gesundheitsanwendungen (DiGA): Das Wichtigste auf einen Blick
06/04/2021: the key points at a glance")
Do you have any questions about the article or would you like to find out more about our services? We look forward to hearing from you!Make a non-binding enquiry now
From our archives (06/04/2021)
Als Hersteller von medizinischen Softwarelösungen möchten Sie sich einen Überblick über das Digitale-Versorgung-Gesetz (DVG) verschaffen und ein besseres Verständnis für die Anforderungen der Digitale-Gesundheitsanwendungen-Verordnung (DiGAV) gewinnen? Katharina Knossalla hat Ihnen alle wichtigen Informationen hier zusammengestellt.Das DVG ist am 19.12.2020 in Kraft getreten und ermöglicht seitdem ca. 73 Millionen gesetzlich Versicherten den Anspruch auf eine Behandlung mittels Digitaler Gesundheitsanwendung (DiGA). Mit dem Gesetz wird es in Zukunft u. a. möglich sein, Online-Sprechstunden einfacher zu nutzen oder sogar ortsunabhängig auf ein sicheres Datennetz im Gesundheitswesen zuzugreifen. Zudem werden derzeit finanzielle Mittel zur Innovationsförderung durch den Bund bereitgestellt. Eigenschaften und Anforderungen an eine DiGA
Was kann eine DiGA sein oder werden? Artikel 2, DVG beschreibt dies genauer. So gelten als DiGA "Medizinprodukte mit niedriger Risikoklasse […], die der Risikoklasse I oder IIa nach Artikel 51 in Verbindung mit Anhang VIII der Verordnung (EU) 2017/745 [über Medizinprodukte] […] zugeordnet und als solche bereits in den Verkehr gebracht sind. […]"Auch bereits auf dem Markt befindliche medizinische Softwarelösungen, welche nach der alten Richtline für Medizinprodukte (MDD) zugelassen sind und vor Inkrafttreten der Verordnung (EU) 2017/745 über Medizinprodukte (Medical Device Regulation - MDR) noch eine Zertifikatsverlängerung erhalten haben, können eine DiGA sein. Bei diesen ist jedoch Vorsicht geboten: Sollte sich im Rahmen einer Zulassung nach besagter Verordnung die Risikoklasse der Software auf Klasse IIb ändern, ist eine Einstufung als DiGA vorerst gesetzlich nicht mehr möglich.Welche Eigenschaften eine DiGA erfüllen muss und was das bedeutet, ist nachfolgend erläutert:| Eigenschaft | Erläuterung |
| Es handelt sich um Software als Medizinprodukt. | Als Medizinprodukt muss die DiGA per Definition des Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM) entweder bei der Erkennung, Überwachung, Behandlung oder Linderung von Krankheiten oder der Erkennung, Behandlung, Linderung oder Kompensierung von Verletzungen oder Behinderungen unterstützen. |
| Die DiGA dient nicht der reinen (Primär-) Prävention. | Auch wenn es sich bei Präventions-Apps prinzipiell um Medizinprodukte handeln kann, sind diese als DiGA Anwendung ausgeschlossen. Wie im DiGA-Leitfaden des BfArM beschrieben, muss es sich um eine digitale Anwendung handeln, welche ein Arzt im Falle einer Krankheitsdiagnose verschreiben kann. |
| Niedrige Risikoklasse nach Anhang VIII der Verordnung (EU) 2017/745 über Medizinprodukte (I oder IIa). | Die 22 risikobasierten Regeln aus dem Anhang der Verordnung müssen gemäß der Zweckbestimmung der medizinischen Software beantwortet werden, um das Risikopotential und damit die Klasse zu bestimmen. Hilfe zur Klassifizierung Ihrer Software finden Sie in der Guidance der Medical Device Coordination Group (MDCG). |
| Die Hauptfunktion beruht auf digitalen Technologien. | Mit einer App bzw. einer browserbasierten Anwendung, einer sog. Stand-alone Software ist dieses Kriterium bereits auf natürliche Weise erfüllt. Mit dieser Anforderung wird Hardware im Allgemeinen (z. B. Blutdruckmessgeräte) als DiGA ausgeschlossen. |
| Der medizinische Zweck wird wesentlich durch die digitale Hauptfunktion erreicht. | Darunter ist zu verstehen, dass Ihre medizinische Softwarelösung nicht nur zum Steuern eines Gerätes oder ausschließlich der Erfassung von Gesundheitsdaten (z. B. mittels eines Sensors) dient, sondern auf Basis von (erhobenen) Daten eine Diagnose oder sogar weiterführende Therapien berechnet. |
| Der Patient ist alleiniger oder Hauptnutzer. | Die DiGA sollte so ausgelegt sein, dass diese vom Patienten allein oder gemeinsam mit dem Arzt verwendet werden kann. Der Arzt darf also bei der Nutzung mit einbezogen, aber nicht der alleinige Nutzer der digitalen Gesundheitsanwendung sein. |
Registrierungsvoraussetzungen für die Listung im BfArM-Verzeichnis
Voraussetzung für eine Listung im Verzeichnis des BfArM ist, dass es sich bei der DiGA um ein CE-gekennzeichnetes Medizinprodukt handelt. Weiterhin existieren folgende Anforderungen, die sich aus der DiGAV ergeben, zu denen es Lösungen zu evaluieren gilt:Anforderungen an Sicherheit und FunktionstauglichkeitDer Nachweis der Sicherheit und Funktionstauglichkeit ihrer medizinischen Software ist durch die Konformitätserklärung des Herstellers bzw. das EG-Zertifikat der Benannten Stelle grundsätzlich als erbracht zu betrachten. Die Basis hierfür bildet die Technische Dokumentation Ihres Medizinprodukts nach Anhang II, der Verordnung (EU) 2017/745 über Medizinprodukte. Diese belegt die Grundlegenden Sicherheits- und Leistungsanforderungen des Anhang I selbiger Verordnung.Anforderungen an Datenschutz und Informationssicherheit
Wichtig ist die Sicherstellung des sorgsamen Umgangs mit Patientendaten, deren Pflege und dem Schutz der Vertraulichkeit, Verfügbarkeit und Integrität. Daher gilt für privatwirtschaftliche Unternehmen neben der Datenschutz-Grundverordnung (DSGVO) auch das Bundesdatenschutzgesetz (BDSG). Zentrale Vorschrift für die Verarbeitung von Gesundheitsdaten ist dabei § 22 BDSG (ggf. i. V. m. Art. 9 DSGVO). Für besondere Bereiche können weitere datenschutzrechtliche Regelungen aus anderen Gesetzen relevant sein (u. a. aus dem Medizinprodukterecht). Anforderungen an Interoperabilität
Interoperabilität bezeichnet die Eigenschaft technischer Systeme auf technisch-syntaktischer, semantischer und organisatorischer Ebene zusammenzuarbeiten. Um die Gesundheitsanwendungen also sinnvoll nutzen zu können, müssen diese perspektivisch miteinander kommunizieren und Informationen austauschen können. Um ein besseres Bild zu bekommen, finden Sie nachfolgend eine Grafik des BfArM (IOP für DiGA). Die Interoperabilitätsschnittstellen, welche Sie in Ihrer medizinischen Softwarelösung bereits zwingend umsetzen müssen, sind unten im Bild in grün dargestellt. Die gepunkteten Linien zeigen, welche Erweiterungen es in Zukunft noch geben soll. Ein Klick auf die Grafik vergrößert sie.
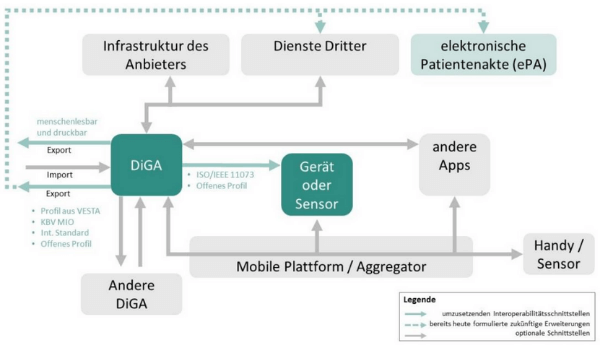
Quelle: Leitfaden des BfArM zum Fast-Track-Verfahren: Transparenter Überblick zum Verfahren und Interpretationshilfe zu den Anforderungen, S. 59, Abb. 7: IOP für DiGA
Anforderungen an Robustheit
Für die Anwender ist es wichtig, dass die Anwendungen ohne Störungen (z. B. Datenverluste, Verbindungsprobleme usw.) genutzt werden können. Des Weiteren sollte beachtet werden, dass fehlerhafte Eingaben von Daten nicht zu Verfälschungen bzw. Einschränkungen des Nutzens der Gesundheitsanwendung führen. Eine Offline-Nutzbarkeit ist bei der DiGA dabei kein Must-have. Es sollte jedoch über umfangreiche Verifikations- und Validierungsaktivitäten sichergestellt sein, dass z. B. bei Abbruch der Internetverbindung keine Daten verloren gehen.Anforderungen an Verbraucherschutz
Als Verbraucher befinden sich Nutzer einer Digitalen Gesundheitsanwendung bereits in einer besonderen Lebens- bzw. Krankheitssituation. Deshalb sollen Hersteller im Rahmen dieser Anforderung Transparenz zum Leistungsumfang der DiGA schaffen. Das beinhaltet u. a., dass auf den Vertriebsplattformen eine genaue Angabe der Zweckbestimmung und der Leistungsmerkmale zu finden sind. Beachten Sie als Hersteller unbedingt, dass DiGAs frei von Werbung sein müssen. Das bedeutet ganz klar, dass keine Werbebanner oder Pop-ups enthalten sein dürfen, weder für Ihre eigenen noch für die Produkte Dritter. Sollten Sie In-App-Käufe bereitstellen, dürfen Sie auf diese zwar sachlich hinweisen, sie aber nicht aktiv bewerben.
Und hier endet die Transparenz noch nicht: Für den Anwender muss ebenfalls ersichtlich sein, welche Anforderungen die DiGA an die Hard- bzw. Software des Nutzers stellt. Für die Aufnahme in das Verzeichnis des BfArM müssen Sie als Hersteller darstellen, für welche mobilen Endgeräte, Webbrowser, Betriebssysteme, zusätzlicher Hardware, etc. Sie die Anwendung ausreichend getestet und freigegeben haben. Nach erfolgreicher Listung im Verzeichnis sind Sie dazu verpflichtet, diese Informationen zu pflegen und entsprechend aktuell zu halten.Anforderungen an Nutzerfreundlichkeit
Bei der Entwicklung von Apps sollten Sie als Hersteller die plattformspezifischen Styleguides kennen und anwenden. Zudem bieten sich Usability-Untersuchungen nach IEC 62366-1 (Norm zur Gebrauchstauglichkeit von Medizinprodukten) an, um eine besonders hohe Nutzerfreundlichkeit nachzuweisen. Um zu bestimmen, ob eine DiGA nutzerfreundlich ist, muss zunächst die angesprochene Zielgruppe definiert werden. Hierbei gilt es zu beachten, dass ggf. ältere Anwender Apps nicht so intuitiv bedienen können wie Jüngere.
Ebenfalls müssen alle im Verzeichnis gelisteten DiGAs seit dem 01.01.2021 entweder Bedienhilfen für Menschen mit Einschränkungen beinhalten oder die durch die Plattform angebotenen Bedienhilfen unterstützen. Diese Bedienhilfen müssen in den Bereichen Sehen, Hören und Motorik so aufgebaut sein, dass es nicht zu Einschränkungen in der Benutzung kommt. Infolgedessen dürfen z. B. verschiedene Schriftgrößen nicht die Lesbarkeit der Anwendung beeinträchtigen. Organisationen wie bspw. die Bundesfachstelle für Barrierefreiheit bieten zu diesem Thema weitere Vorgaben und Hinweise, an denen Sie sich orientieren können. Sollte es aus der Zweckbestimmung bzw. Ihrer Zielgruppe heraus begründbar sein, können hierbei Ausnahmen zutreffen. Anforderungen an die Unterstützung der Leistungserbringer
Wie bereits beschrieben legen Sie für Ihre DiGA eine Zweckbestimmung und einen positiven Versorgungseffekt fest. Hierbei können neben dem behandelnden Arzt und dem Versicherten noch weitere Leistungserbringer eine Rolle spielen. Mit der Definition von Nutzerrollen beschreiben Sie als Hersteller, welcher Rolle im Gesamtkontext der Behandlung welche Aufgaben zugeschrieben werden. Darauf aufbauend wird geraten, einen prototypischen Ablauf des Versorgungsszenarios zu gestalten, damit der Leistungserbringer dem Versicherten das Zusammenspiel der Nutzerrollen erklären und die Nutzung der DiGA im Rahmen der Therapie erläutern kann. Bei der Antragstellung zur Listung im BfArM-Verzeichnis müssen Sie als Hersteller diese Informationen vorlegen (gemäß § 2 Absatz 1 Nummer 16 DiGAV).Anforderungen an die Qualität der medizinischen Inhalte
Alle präsentierten Inhalte der DiGA müssen sich auf medizinisch-fachliche Informationen, die aus akzeptierten und belastbaren Quellen stammen, stützen. Als Hersteller eines Medizinprodukts können Sie hier auf Ihre Klinische Bewertung und den darin definierten klinischen Nutzen verweisen. Die Klinische Bewertung ist Teil Ihrer Technischen Dokumentation und hat zum Ziel, die Sicherheit und Leistung Ihrer DiGA im klinischen Umfeld zu bewerten. Dazu bedient man sich Daten aus einschlägiger wissenschaftlicher Literatur und/oder klinischen Prüfungen. Diese Daten werden gesammelt, analysiert und bewertet. Da es sich bei der Klinischen Bewertung um ein methodisch fundiertes und fortlaufendes Verfahren handelt, muss diese in einem vom Hersteller festgelegten Zeitraum aktualisiert werden.
Achten Sie bei der Darstellung von medizinisch-fachlichen Informationen auch im Rahmen der Usability Ihres Produkts darauf, dass die Informationen angemessen und für Ihre definierte Zielgruppe verständlich formuliert und abgebildet sind. Anforderungen an die Patientensicherheit
Mit Vergabe des CE-Kennzeichens, und damit verbunden einem Risikomanagement nach ISO 14971, gilt die technische Sicherheit Ihres Medizinproduktes als grundsätzlich gewährleistet. Im Rahmen der Patientensicherheit sollten jedoch ebenfalls pathologische Zustände des Versicherten, die hinlänglich ihrer klinischen Risiken bekannt sind, betrachtet werden. DiGAs werden z. T. bei risikobehafteten Zuständen des Versicherten angewandt. Für Diabetiker wäre es z. B. wichtig, innerhalb der Anwendung auf das Risiko und die möglichen Folgen einer Unterzuckerung hingewiesen zu werden.
Mit der Anforderung an die Patientensicherheit soll der Hersteller den Anwender der DiGA für bekannte Restrisiken (z. B. mittels eines Warnhinweises) sensibilisieren und sicherzustellen, dass der Anwender eigenständig erkennt, wann Rücksprachen mit einem Arzt oder evtl. sogar ein Abbruch der Anwendung erfolgen muss. Abhängig vom bewerteten Restrisiko für die Patientensicherheit besteht für Sie als Hersteller eventuell die Möglichkeit, Warnhinweise über eine Check-Box in den Einstellungen vom Versicherten ausschalten zu lassen.
Fazit
Wie Sie sehen sind die Anforderungen an Digitale Gesundheitsanwendungen hoch, aber nicht unüberwindbar. Von Datenschutz über Interoperabilität bis hin zu Robustheit und Qualität an medizinisch-fachliche Informationen: Jetzt haben Sie schwarz auf weiß, für welche Anforderungen Sie Ihre Technische Dokumentation zum Nachweis nutzen können. Seit 20 Jahren unterstützt Metecon Hersteller von Medizinprodukten bei der Erstellung und Pflege ihrer Technischen Dokumentationen und sorgt damit für die Zulassung sicherer und leistungsfähiger Produkte. Profitieren Sie von unserer Erfahrung und lassen Sie sich von unseren Experten beraten und unterstützen: bei der Klinischen Bewertung, beim Risikomanagement, bei der Usability oder der Verifikation/Validierung für die Erfüllung der IEC 62304.Kontaktieren Sie uns. Wir freuen uns, von Ihnen zu hören!
Our blog posts are researched and created with the utmost care, but are only snapshots of the regulations, which are constantly changing. We do not guarantee that older content is still current or meaningful. If you are not sure whether the article you have read on this page still corresponds to the current state of regulation, please contact us: we will quickly place your topic in the current context.



