
IVDR-Proposal der EU-Kommission: Neue Übergangsfristen
29/10/2021
Do you have any questions about the article or would you like to find out more about our services? We look forward to hearing from you!Make a non-binding enquiry now
From our archives (29/10/2021)
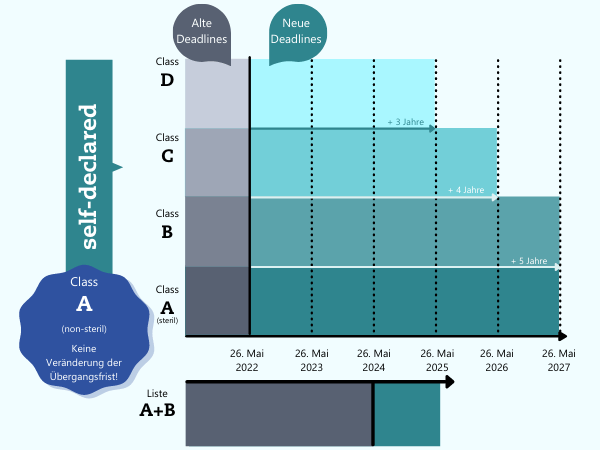
Die EU-Kommission hat im Oktober 2021 neue Übergangsfristen für die Einhaltung der EU-IVDR vorgeschlagen. Damit soll dem Mangel an IVDR-benannten Benannten Stellen begegnet werden. Auch den IVD-Herstellern kommt diese Lösung entgegen, weil ihnen unter bestimmten Bedingungen so etwas mehr Zeit für die Anpassung verschafft wird:"Um dem Mangel an Kapazitäten der benannten Stellen auf breiterer Basis zu begegnen, schlägt die Kommission zusätzliche Übergangsfristen für Produkte vor, die zum ersten Mal im Rahmen der IVD-Verordnung einer Konformitätsbewertung durch benannte Stellen unterzogen werden müssen. Der Vorschlag unterscheidet zwischen Risikoklassen und sieht eine Übergangsfrist bis Mai 2025 für Produkte mit hohem Risiko (Klasse D), bis Mai 2026 für Produkte der Klasse C und bis Mai 2027 für Produkte mit geringerem Risiko (sterile Produkte der Klassen B und A) vor. Dieser Ansatz zielt darauf ab, ein Gleichgewicht zwischen der verfügbaren Kapazität der benannten Stelle und dem hohen Schutzniveau der öffentlichen Gesundheit herzustellen." (Zitat: EU-Kommission im Q&A-Teil)Wer profitiert von den Neuerungen?
Für IVD-Hersteller, welche bereits ein IVDD-Zertifikat für Ihre IVDs haben (ca. 8 % nach Schätzung der Europäischen Kommission in Brüssel) wird die ursprünglich geplante Übergangszeit um ein Jahr verlängert, von Mai 2024 auf Mai 2025. Hersteller sogenannter Inhouse-Tests (LDTs) profitieren ebenfalls von einer um zwei bzw. vier Jahren verlängerten Übergangsfrist: Mai 2024, bzw. Mai 2028. Die erweiterte Frist bis 2028 betrifft die Anforderungen an das Fehlen äquivalenter Produkte, mehr dazu in diesem Artikel.Hersteller sonstiger Produkte (self-declared, ca. 80 %), deren Produkte in Zukunft in die Klassen D, C, B oder A – steril fallen, profitieren von den neuen Übergangsfristen.
PDF-Grafik hier downloaden
Was bleibt unverändert?
Sicher ist: Die IVDR tritt am 26.05.2022 in Kraft, Änderungen hin oder her.Insbesondere gilt die IVD-Verordnung ab dem 26. Mai 2022 in vollem Umfang für CE-gekennzeichnete In-vitro-Diagnostika, die ohne Mitwirken von Benannten Stellen legal auf dem EU-Markt in Verkehr gebracht werden dürfen (d. h. nicht-sterile Produkte der Klasse A, die etwa 20 % des Marktes ausmachen), und für "neue" In-vitro-Diagnostika (d. h. Produkte, für die keine vor dem 26. Mai 2022 ausgestellte Bescheinigung oder Konformitätserklärung des Herstellers vorliegt).Produkte, die von den (zusätzlichen) Übergangsfristen betroffen sind, können diese nur in Anspruch nehmen, wenn sie bestimmte Bedingungen erfüllen, insbesondere, dass sie weiterhin der IVDD entsprechen und dass es keine wesentlichen Änderungen an ihrer Auslegung und Zweckbestimmung gibt.Darüber hinaus gelten ab diesem Zeitpunkt die verschärften Vorschriften für Vigilanz und Marktüberwachung auch für Produkte, für die die Übergangsfristen gelten.
Tipps zum Umgang mit möglichen Konflikten
IVD-Hersteller haben sich bereits mit der Bitte um Beratung an uns gewandt: Sie sollten eigentlich von dem Vorschlag der Kommission profitieren, haben jedoch keinen gültigen Performance Evaluation Report (PER, Leistungsbewertungsbericht) vorliegen, da dieser unter der Richtlinie noch nicht gefordert wurde. Diese Hersteller sind also nicht in der Lage, die geforderten Vorschriften der Marktüberwachung aus Artikel 110 (3) vollständig zu erfüllen, da die Aufnahme von Post-Market Performance Follow-Up oder eine begründete Ablehnung von PMPF durch ein Non-PMPF-Statement den PER voraussetzt. Da PMPF aus der Performance Evaluation/Leistungsbewertung resultiert, gibt es keine Fortsetzung ohne initiale Leistungsbewertung, welche aber erst nach der verlängerten Übergangsfrist notwendig zu haben ist!Folgende Möglichkeiten sehen wir, um diesen Konflikt zu lösen:
- Möglichkeit 1: Sie hinterlegen ein Non-PMPF-Statement im PMS-Plan, welches darauf hinweist, dass bis zur Erstellung des PERs kein PMPF möglich ist.
- Möglichkeit 2: In Ihrem PMPF-Plan veranschlagen Sie, die Notwendigkeit für PMPF mit Erhalt/Fertigstellung des PERs zum gegebenen Zeitpunkt zu bewerten.
- Möglichkeit 3: Sie erstellen einen PER, indem Sie das Ableiten von PMPF zur Notwendigkeit erklären.
Our blog posts are researched and created with the utmost care, but are only snapshots of the regulations, which are constantly changing. We do not guarantee that older content is still current or meaningful. If you are not sure whether the article you have read on this page still corresponds to the current state of regulation, please contact us: we will quickly place your topic in the current context.

Regulatory Affairs Expert




