
Als Hersteller von Medizinprodukten sind Sie aufgefordert, aktive Marktbeobachtung zu betreiben und definierte Post-Market Surveillance (PMS)-Strategien für jedes Produkt bzw. jede Produktfamilie zu erarbeiten. Das versetzt Sie in die Lage, unterschätzte Komplikationen und Probleme, die vor der Markteinführung nicht zu erkennen waren, sowie unvorhergesehenen Gebrauch Ihres Produkts über den gesamten Produktlebenszyklus zu erkennen und zu beheben.Zu den Aktivitäten für die Post-Market Surveillance können zählen: die Bearbeitung von Reklamationen; Vigilanz; die Befragung von Patienten, Anwendern, Händlern und Transporteuren; die Auswertung von Rückmeldungen aus der Hotline, aus Social Media-Foren bis hin zu Bewertungen aus dem App-Store; Recherche in Datenbanken der Überwachungsbehörden zu Vorkommnissen und Rückrufen zu ähnlichen Produkten auf dem Markt sowie produktspezifische Post-Market Clinical Follow-up-Maßnahmen (PMCF-Maßnahmen). Dabei zählt jede Aktivität, die nicht anlassbezogen ist, als aktiv.Durch eine gemeinsam erarbeitete Strategie können Sie diese Daten effizient in Ihre Klinische Bewertung einbringen und sparen so bei der (jährlichen) Aktualisierung viel Zeit und hohe Personalkosten.Entwickeln Sie mit uns eine PMS-Strategie, die praxisnah, effizient und MDR-konform ist – für mehr Sicherheit und weniger Aufwand.
Definition Post-Market Surveillance (PMS)
Post-Market Surveillance (PMS) bezeichnet einen systematischen Prozess des Herstellers für die Überwachung nach dem Inverkehrbringen. Wie und wozu es PMS braucht, wird in Kapitel 7 "Überwachung nach dem Inverkehrbringen, Vigilanz und Marktüberwachung" der MDR (EU-Verordnung 2017/745) erläutert. ISO 13485:2016 und ISO 14971:2022 fordern ebenfalls eine Überwachung nach dem Inverkehrbringen.Unsere PMS-Services
Leistung sichern & Risiken minimieren mit der richtigen PMS-StrategieDie Überwachung nach dem Inverkehrbringen ist für Medizinprodukte nach MDR verpflichtend und entscheidend für die kontinuierliche Bewertung von Sicherheit, Leistung und klinischem Nutzen. Wir unterstützen Sie mit individuellen PMS-Strategien, die regulatorische Anforderungen nicht nur erfüllen, sondern auch Effizienzpotenziale in Ihrem Unternehmen sichtbar machen.Aktuelle Anforderungen immer im Blick ohne Mehraufwand:
Unser Live-PMS-Ticker hält Sie über nationale und internationale Entwicklungen auf dem Laufenden. So reagieren Sie frühzeitig auf regulatorische Änderungen.Digitale Tools für ein PMS-System, das mitdenkt:
Wir begleiten Sie bei der Auswahl und Integration digitaler Lösungen, mit denen Sie Ihren PMS-Prozess optimieren und langfristig Ressourcen sparen.Stärker im Team durch praxisorientiertes Know-how:
Wir qualifizieren Ihre Teams gezielt zu MDR-konformer PMS und zeigen, wie Sie strukturiert und effizient mit Rückmeldungen, Reklamationen oder wissenschaftlicher Literatur umgehen.Mehr Länder, gleiche Strategie: internationale Skalierbarkeit:
Ihr bestehendes PMS-System erweitern wir gezielt für internationale Märkte, ohne es vollständig umzustellen. Das spart Aufwand und erhöht die Konsistenz in Ihrer Marktbeobachtung.Maßgeschneiderte PMS-Konzepte für Ihre Produkte:
Wir entwickeln PMS-Strategien, die zu Ihrer Produktarchitektur und Ihren internen Abläufen passen. So bleibt der Aufwand planbar und die Aussagekraft hoch.Trends erkennen, bevor sie kritisch werden:
Durch systematische Auswertung Ihrer PMS-Daten und Rückmeldungen identifizieren wir frühzeitig Auffälligkeiten und leiten daraus konkrete Handlungsoptionen ab.Dokumente, die Ihnen wirklich etwas bringen:
Wir erstellen Ihre PMS-Pläne, PMS-Berichte und PMPF-Dokumente so, dass sie regulatorisch belastbar sind und zugleich als wertvolles Steuerungsinstrument dienen.Sicher auftreten gegenüber Behörden und Benannten Stellen:
Wir begleiten Sie bei Audits, Rückfragen und Bewertungen durch Aufsichtsbehörden: strukturiert, nachvollziehbar und immer auf Augenhöhe.
Kontaktieren Sie uns, und wir finden gemeinsam heraus, wie wir Sie unterstützen können.
Unser Live-PMS-Ticker hält Sie über nationale und internationale Entwicklungen auf dem Laufenden. So reagieren Sie frühzeitig auf regulatorische Änderungen.Digitale Tools für ein PMS-System, das mitdenkt:
Wir begleiten Sie bei der Auswahl und Integration digitaler Lösungen, mit denen Sie Ihren PMS-Prozess optimieren und langfristig Ressourcen sparen.Stärker im Team durch praxisorientiertes Know-how:
Wir qualifizieren Ihre Teams gezielt zu MDR-konformer PMS und zeigen, wie Sie strukturiert und effizient mit Rückmeldungen, Reklamationen oder wissenschaftlicher Literatur umgehen.Mehr Länder, gleiche Strategie: internationale Skalierbarkeit:
Ihr bestehendes PMS-System erweitern wir gezielt für internationale Märkte, ohne es vollständig umzustellen. Das spart Aufwand und erhöht die Konsistenz in Ihrer Marktbeobachtung.Maßgeschneiderte PMS-Konzepte für Ihre Produkte:
Wir entwickeln PMS-Strategien, die zu Ihrer Produktarchitektur und Ihren internen Abläufen passen. So bleibt der Aufwand planbar und die Aussagekraft hoch.Trends erkennen, bevor sie kritisch werden:
Durch systematische Auswertung Ihrer PMS-Daten und Rückmeldungen identifizieren wir frühzeitig Auffälligkeiten und leiten daraus konkrete Handlungsoptionen ab.Dokumente, die Ihnen wirklich etwas bringen:
Wir erstellen Ihre PMS-Pläne, PMS-Berichte und PMPF-Dokumente so, dass sie regulatorisch belastbar sind und zugleich als wertvolles Steuerungsinstrument dienen.Sicher auftreten gegenüber Behörden und Benannten Stellen:
Wir begleiten Sie bei Audits, Rückfragen und Bewertungen durch Aufsichtsbehörden: strukturiert, nachvollziehbar und immer auf Augenhöhe.
Kontaktieren Sie uns, und wir finden gemeinsam heraus, wie wir Sie unterstützen können.
Mit smarten Updates Zeit und Kosten sparen
Eine Klinische Bewertung von MDD auf MDR anzupassen, ist meist mit größerem Aufwand verbunden. Ist eine gute MDR-taugliche Grundlage geschaffen und sind die dazugehörigen PMCF-Maßnahmen optimal definiert, lassen sich die Updates deutlich schneller und damit kostengünstiger erstellen.
Lassen Sie uns gemeinsam und unverbindlich herausfinden, wie wir Ihre Klinische Bewertung und Ihre Post-Market Surveillance optimieren können. Wir freuen uns, von Ihnen zu hören!
Unser Angebot:
Wir kümmern uns vollumfänglich um Ihre Klinische Bewertung. Dazu erinnern wir Sie auch daran, wann ein Update ansteht, holen die entsprechenden Informationen ein und führen das Update durch.Ihre Vorteile:
Die bevorstehenden Updates laufen über die PMCF-Maßnahmen, die wir zuvor im Rahmen Ihrer Klinischen Bewertung schlau und effizient definiert haben. Wir halten Ihren Aufwand so gering wie möglich, und Sie bekommen Ihre Klinische Bewertung immer aus einer Hand.Lassen Sie uns gemeinsam und unverbindlich herausfinden, wie wir Ihre Klinische Bewertung und Ihre Post-Market Surveillance optimieren können. Wir freuen uns, von Ihnen zu hören!
Welche Bedeutung hat PMS für Ihre Medizinprodukte?
Welche Bedeutung PMS für Ihre Medizinprodukte hat, lässt sich aus verschiedenen Blickwinkeln betrachten:- Die Post-Market Surveillance ist Bestandteil des Qualitätsmanagements (inbes. des Risikomanagements) von Medizinprodukten in Europa.
- Sie dient der Überwachung, der Sicherheit und der Leistung von Medizinprodukten nach ihrer Markteinführung.
- Informationen über die Verwendung von Medizinprodukten werden gesammelt und analysiert, um Risiken oder Probleme im Feld frühzeitig zu erkennen und Maßnahmen einzuleiten.
- Die Post-Market Surveillance trägt zum Schutz von Patienten und Anwendern bei und hilft dabei, dass Medizinprodukte sicher und wirksam bleiben.
Ihre Anforderungen an PMS – unsere Lösungen
Unser Team erarbeitet mit Ihnen Ihre PMS-Strategie, die Ihre Anforderungen erfüllt, und erstellt alle grundlegenden Dokumente wie den PMS-Prozess, die PMS-Pläne und die Berichtsvorlagen.Profitieren Sie von unserem Quick-Check und finden Sie schnell heraus, welche Unterstützung Sie benötigen!Überwachung nach dem Inverkehrbringen (PMS) für Medizinprodukte unter der MDR
Die Überwachung nach dem Inverkehrbringen (Post-Market Surveillance, PMS) ist mittlerweile sowohl nach MDR als auch nach IVDR vorgeschrieben. Wahrscheinlich haben Sie bereits praktische Erfahrungen mit den Herausforderungen, die PMS nach MDR mit sich bringen. Vielleicht kennen Sie bereits eines der Szenarien:- Die PMS erfordert mehr Ressourcen als erwartet.
- Ihr Team ist noch nicht fit genug, um mit den Herausforderungen zurecht zu kommen, die die beiden Verordnungen an sie stellen.
- Bei Ihrem letzten Audit gab es Neben- oder Hauptabweichungen.
Wir wollen Teil der Lösung für Ihre PMS-Herausforderungen sein. Finden Sie mit nachfolgenden Quick-Check schnell heraus, was Sie brauchen und wie wir Sie dabei unterstützen können!
Quick-Check
So profitieren Sie von unserer Expertise in Post-Market Surveillance
Unserer Erfahrung nach stellt PMS Sie auf fünf unterschiedliche Arten vor große Herausforderungen. Ordnen Sie sich nachfolgend einem Profil zu und sehen Sie, welche Lösung wir Ihnen anbieten. Klingt interessant? Darüber sollten wir uns einmal unverbindlich austauschen?Wir freuen uns, von Ihnen zu hören!
1. Kick Start
Sie brauchen eine sichere PMS-Basis und eine durchdachte Strategie
Sie sind sich der Anforderungen an PMS bewusst und stehen am Anfang Ihrer PMS-Einführung? Oder wollen Sie mit Grundlagen starten und brauchen PMS-Strategien für die Implementierung? Kein Problem, wir helfen Ihnen loszulegen!Unser Ansatz:Wir stellen Ihr Unternehmen nicht auf den Kopf. Unser Ziel ist es, eine regulatorisch konforme und umsetzbare Strategie für Ihr Unternehmen zu entwickeln. Um auf der gleichen Wellenlänge zu sein, müssen wir Sie und Ihr Produktportfolio kennenlernen und die Ist-Situation aufnehmen. In einem gemeinsamen Workshop erstellen wir Ihnen einen individuellen Strategieentwurf. Zum Schluss führen wir (gemeinsam) Ihr PMS mit der ausgearbeiteten Strategie ein und messen ihre Wirksamkeit.Wir bringen Sie sicher ins Ziel - Jetzt mehr erfahren!

2. Fast Lane
Sie müssen Gas geben
Hilfe, schnell! Sie haben eine Abweichung in Ihrem PMS und brauchen jetzt eine zupackende Hand, um schnell wieder die Konformität herzustellen. Gemeinsam bringen wir alles wieder auf Kurs!Unser Ansatz:Wenn Sie schnell handeln müssen, zählt Erfahrung! Unsere Expert*innen wissen, wie eine Nichtkonformität zeitnah zu beheben ist. Wir sind uns bewusst, dass Sie unter Druck stehen und dass die Uhr tickt. Gemeinsam finden wir zügig die optimale Lösung, um Ihre Abweichung zu beheben. Mit uns haben Sie einen zuverlässigen Partner für schwierige Zeiten an Ihrer Seite.Wir sind bereit für Ihren Boxenstopp!

3. Power, Power, Power!
Sie sind ein Tuning-Fan
Sie haben ein etabliertes PMS-System, allerdings fehlt Ihnen die Power, um alle Aufgaben angemessen und rechtzeitig zu erledigen? Unser Team ist für Sie da!Unser Ansatz:Mit zu wenigen Ressourcen kann auch das beste PMS-System nicht richtig funktionieren. Wir stellen Ihnen die in solchen Zeiten benötigten Kapazitäten zur Verfügung und helfen Ihnen, alle Anforderungen einzuhalten. Dank unserer Erfahrung arbeiten wir uns schnell in die Materie ein und erledigen die anfallenden Aufgaben zuverlässig.Verstärkung fürs Rennteam gesucht? Wir freuen uns aufs Kennenlernen!

4. Saving Energy
Sie stehen auf effiziente Prozesse
Sie benötigen mit dem etablierten PMS-System zu viele interne Kapazitäten (personell, zeitlich, finanziell). Wir helfen Ihnen, Ihre Prozesse effizienter zu gestalten.Unser Ansatz:Für uns ist Energiesparen mehr als der Einsatz von Software. Wir wissen, dass es herausfordernd ist, Ressourcen zu optimieren und gleichzeitig so energieeffizient wie möglich zu sein. Wir schauen uns gemeinsam mit Ihnen als objektive Dritte Ihre PMS-Prozesse an, um Ihre Energieeffizienz zu verbessern und gleichzeitig die MDR-Vorschriften einzuhalten. Selbst kleine Änderungen führen im Laufe der Zeit zu großen Einsparungen!Schonen Sie Ihren Motor mit unserer Unterstützung!

5. Driver´s License
Ihr Team braucht ein PMS-Update
Sie haben interne Kapazitäten, welche entsprechend ausgebildet werden müssen. Kein Problem, unsere Trainer*innen geben ihr Wissen gerne an Sie weiter.Unser Ansatz:Im Rahmen der Metecon Academy bieten wir Ihnen maßgeschneiderte Fortbildungen fürs gesamte Team an. Wir verschaffen uns gemeinsam mit Ihnen eine Übersicht des aktuellen Know-Hows. Unsere Trainer*innen sind nicht nur Expert*innen in ihrem Fach – Ihr Erfolg liegt uns am Herzen! Am Ende unserer Workshops erarbeiten wir gemeinsam einen Probelauf an einem Ihrer Produkte, um die Wirksamkeit der Trainingsinhalte in der Praxis sicherzustellen.Weitere Infos liefern wir gerne!

Sie sehen einen anderen Bedarf für sich?
Was uns motiviert


Das Produkt ist am Markt, die Klinische Bewertung ist gelaufen?
Nein, denn als Hersteller müssen Sie über die gesamte Produktdauer die Sicherheit und Leistungsfähigkeit Ihres Produkts garantieren können. Um geeignete Post Market Clinical Follow-Up (PMCF)-Aktivitäten im Rahmen der Post-Market Surveillance (PMS) festzulegen, gibt es verschiedene Wege, wie die Auswertung bestimmter Fragestellungen mit großer Patientenzahl, Langzeit-Beobachtungen von Patienten oder Untersuchungen zu bekannten Risiken aus der Literatur. Anhand dieser Daten erkennen Sie etwaige Probleme Ihres Produkts, beseitigen diese frühzeitig und passen Ihre Nachfolgeprodukte immer weiter an den Bedarf Ihrer Anwender an.Post-Market Clinical Follow-Up (PMCF)
Mögliche PMCF-Maßnahmen umfassen Recherche in wissenschaftlicher Literatur und in Vorkommnis-Datenbanken sowie Umfragen bei Patienten und Anwendern zum Produkt. Post-Market Clinical Follow-Up (PMCF)-Studien sind sogenannte Marktbeobachtungsstudien, die für Medizinprodukte mit rechtmäßigem CE-Kennzeichen durchgeführt werden. Dabei muss das Medizinprodukt innerhalb seiner Zweckbestimmung eingesetzt sein, das Studiendesign darf weder zusätzliche invasive noch zusätzlich belastende Untersuchungen beinhalten. Diese Studien dienen dazu, weitere klinische Daten zu generieren und diese in den klinischen Bewertungsbericht einfließen zu lassen. Da die ISO 14155 keine Unterscheidung zwischen Medizinprodukten mit und ohne CE-Kennzeichnung macht, ist diese Norm für alle Studien/Klinischen Prüfungen gleichermaßen anzuwenden.Wir bringen Struktur in Ihre internen Prozesse der Marktbeobachtung und zeigen Ihnen Möglichkeiten auf, wie Sie diese Daten, gerne auch mit unserer Unterstützung, effizient in die Klinische Bewertung einarbeiten. Wenn die Ergebnisse zeigen, dass Sie eine PMCF-Studie durchführen müssen, planen wir diese Studie effizient und sorgen für ihren reibungslosen Verlauf. Insbesondere bei der Erstellung des PSUR oder PMS-Reports stehen wir Ihnen gerne zur Seite – wir wissen, dass diese für Hersteller oft herausfordernd sind.Sie wollen mehr erfahren? Unsere Erstgespräche sind natürlich kostenfrei!MDR: Product Lifecycle Reporting
Mit der MDR kommen neue Verpflichtungen auf Sie als Medizinprodukte-Hersteller zu, wie das regelmäßige Anfertigen von Plänen und Berichten. Manche Aktualisierungszeitpunkte sind dabei fest vorgeschrieben, andere müssen in diesen Zyklus sinnvoll eingegliedert werden.Das Schaubild stellt die Pläne und Berichte in logischer Abfolge dar. So fällt es leichter, Abhängigkeiten zu erkennen. Bereits während der Produktentwicklung muss die Gewinnung klinischer Daten geplant werden. Nach der Zulassung befindet man sich über den gesamten Produktlebenszyklus in einem fortlaufenden Prozess der Anfertigung von Plänen und Berichten. Um allen Anforderungen gerecht zu werden und alle Dokumente rechtzeitig bereitstellen zu können, müssen die einzelnen Phasen gut aufeinander abgestimmt sein.
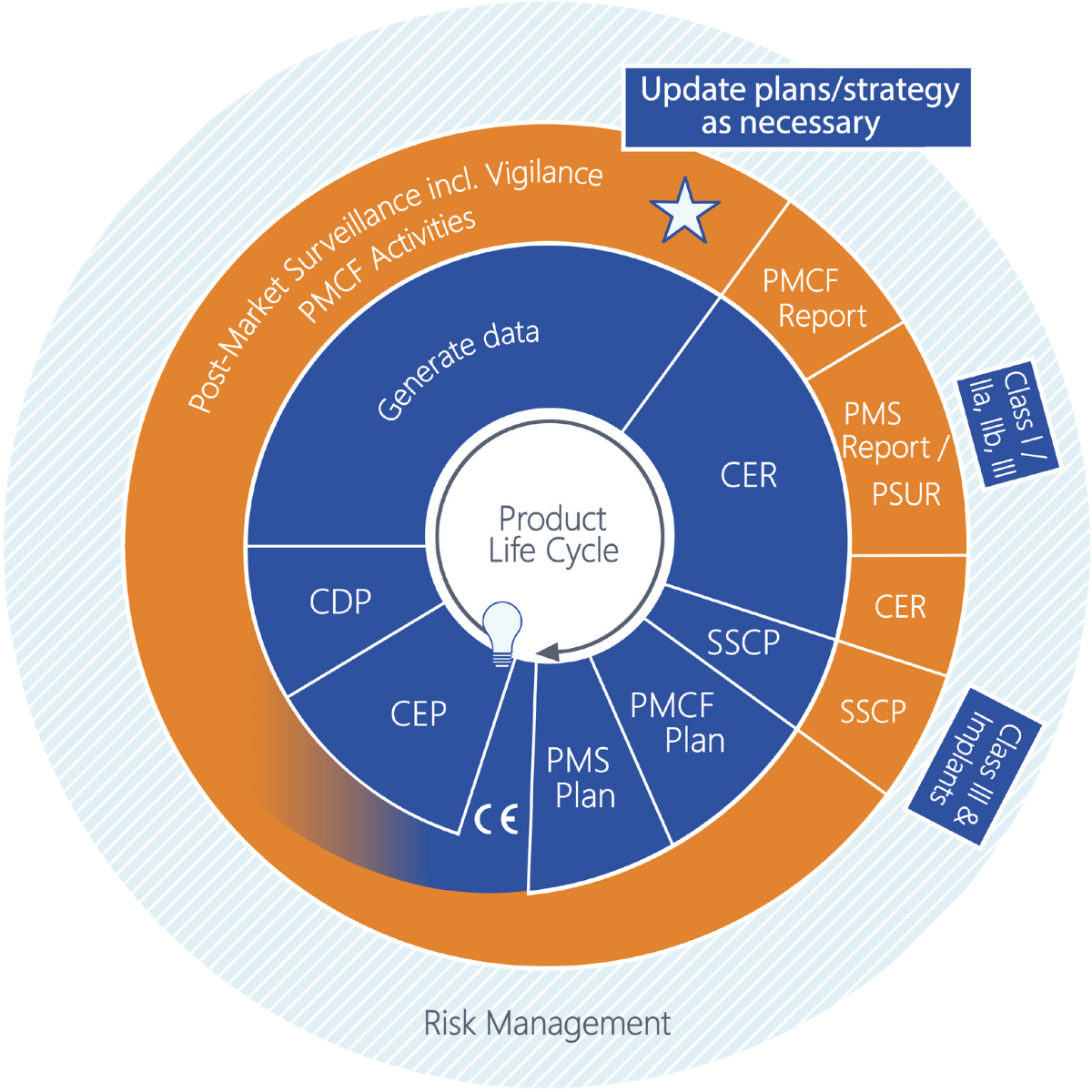
Erläuterungen zur Grafik
+CEP – Clinical Evaluation Plan (Klinischer Bewertungsplan)
Der CEP definiert den Umfang der klinischen Evidenz. Er legt die Aspekte des bestimmungsgemäßen Verwendungszwecks (und damit die beabsichtigte klinische Leistung und den klinischen Nutzen) sowie die Restrisiken fest. Die Parameter, die zur Bewertung des Nutzen-Risiko-Verhältnisses erforderlich sind, werden identifiziert und alle Marketingaussagen müssen durch geeignete Daten belegt werden. Die Strategie (der Weg) der klinischen Bewertung wird unter Berücksichtigung dieser Aspekte und Parameter, bereits vorliegender präklinischer und klinischer Daten, der Risikoklasse und möglicher gleichwertiger Produkte festgelegt. Schließlich wird die Suchstrategie definiert, einschließlich mindestens einer Literatursuche (Planung der Quellen, Suchbegriffe, Auswahl- und Bewertungskriterien) und, falls erforderlich, einer klinischen Studie. Der CEP wird aktualisiert, wenn sich Umfang oder Vorgehen ändern.
+CDP – Clinical Development Plan (Klinischer Entwicklungsplan)
Der CDP definiert, wie die erforderlichen klinischen Daten generiert werden. Dies kann explorative Studien, First-in-Man-Studien, Machbarkeitsstudien und Pilotstudien bis hin zu Proof-of-Concept-Studien umfassen. Auch ein Ausblick auf mögliche PMCF-Aktivitäten ist an dieser Stelle möglich. Der CDP wird aktualisiert, wenn sich Studienziele ändern oder zusätzliche Studien hinzukommen.
+CER – Clinical Evaluation Report (Klinischer Bewertungsbericht)
Der CER stellt die Ergebnisse der Bewertung aller klinischen Daten dar. Daten aus dem Produkt oder aus nachweislich gleichwertigen Produkten werden gesammelt, ausgewählt, bewertet und analysiert. Es wird geprüft, ob und in welchem Umfang die Anforderungen an Leistung/Nutzen und Sicherheit (Risiken und unerwünschte Nebenwirkungen) erfüllt werden und ob das resultierende Nutzen-Risiko-Verhältnis akzeptabel ist. Der CER benennt zudem den Bedarf an weiteren klinischen Daten im Rahmen von PMCF. Er wird kontinuierlich mit PMCF-Daten aktualisiert, um die fortwährende Konformität mit den GSPR nachzuweisen.
+PMCF-Plan – Post-Market Clinical Follow-up Plan
PMCF ist formaler Bestandteil der PMS und dient der Erweiterung der klinischen Evidenz zum Produkt über den gesamten Lebenszyklus. PMCF beantwortet zudem offene Fragen, die bislang nur im Rahmen der klinischen Bewertung geschätzt werden konnten (z. B. Langzeitverhalten, Überwachung von Nebenwirkungen und Kontraindikationen). Der PMCF-Plan beschreibt die Methoden und Verfahren zur proaktiven Sammlung oder Generierung klinischer Daten. Der Umfang kann je nach erforderlichen PMCF-Aktivitäten variieren; ggf. wird ein PMCF-Masterplan erstellt, der auf verschiedene andere Pläne verweist, in denen die einzelnen Aktivitäten definiert sind. Der PMCF-Plan wird aktualisiert, wenn dies erforderlich ist – beispielsweise wenn im PMCF-Bericht ein Änderungsbedarf festgestellt wurde oder wenn der CER geänderte oder zusätzliche PMCF-Anforderungen identifiziert hat.
+PMS-Plan – Post-Market Surveillance Plan
Im PMS-Plan definiert der Hersteller, wie das Produkt nach dem Inverkehrbringen überwacht wird, einschließlich der zu erhebenden Daten. Bei der produktspezifischen Planung dieser Aktivitäten sind die Eigenschaften und die damit verbundenen Risiken des Produkts sowie die Erkenntnisse aus dem CER zu berücksichtigen. Darüber hinaus werden die Methoden zur Datenauswertung und die Bewertungskriterien festgelegt. Die Datenerhebung auf dem Markt muss proaktiv erfolgen. Der PMS-Plan ist zu aktualisieren, wenn neue Aspekte zu berücksichtigen sind (z. B. ein neuer klinischer Nutzen), wenn Verbesserungsmöglichkeiten identifiziert werden oder wenn sich die Produkte ändern. Ebenso muss er aktualisiert werden, wenn neue Datensätze verfügbar werden, die in die PMS aufgenommen werden sollten.
+PMS- (einschließlich Vigilanz) und PMCF-Aktivitäten
Bei der Planung proaktiver PMS-Aktivitäten ist zu berücksichtigen, welche Aktivitäten als PMCF-Aktivitäten durchgeführt werden sollen, da nicht jede proaktive PMS-Aktivität die Kriterien für PMCF erfüllt. Typische proaktive PMS- oder PMCF-Datensätze sind Recherchen in der wissenschaftlichen Literatur und veröffentlichte Berichte über klinische Erfahrungen mit dem eigenen Produkt oder mit gleichwertigen Produkten. Informationen zu ähnlichen Produkten werden auch im Rahmen einer Suche in Datenbanken zu Vorkommnissen oder FSCA (Field Safety Corrective Actions) erhoben. Anwenderbefragungen können je nach Zielsetzung entweder als proaktive PMS- oder als PMCF-Aktivität durchgeführt werden.Reaktive Datensätze (Teil der PMS) können z. B. schwerwiegende Vorkommnisse, FSCAs und Trendberichte umfassen. Informationen und Ereignisse werden unmittelbar nach ihrem Eingang bewertet, und die entsprechenden Folgemaßnahmen werden sofort eingeleitet. In diesem Zusammenhang bezeichnet Vigilanz die Meldung schwerwiegender Vorkommnisse und FSCAs an die Behörde. Hierfür benötigt jeder Hersteller ein geeignetes System, das die Bewertung und Analyse solcher Ereignisse sowie deren fristgerechte Meldung sicherstellt. Die Meldung erfolgt über EUDAMED.
+PMCF-Bericht – Post-Market Clinical Follow-up Report
Die klinischen Daten und ggf. Zwischenergebnisse aus PMCF-Aktivitäten werden im PMCF-Bericht zusammengefasst und dargestellt. Die Daten werden hinsichtlich der Bestätigung der klinischen Bewertung sowie der Wirksamkeit der Aktivitäten geprüft. Der PMCF-Bericht kann auch über Folgemaßnahmen entscheiden. Er wird regelmäßig entsprechend den im PMS festgelegten Datenerhebungszeiträumen aktualisiert.
+PMS-Bericht und Periodischer Sicherheitsbericht (PSUR)
Für Produkte der Klasse I wird ein PMS-Bericht erstellt. Er enthält eine Zusammenfassung der Ergebnisse aus PMS-Daten (einschließlich PMCF-Daten), die im Erhebungszeitraum gewonnen wurden. Auf Basis dieser Ergebnisse wird ein Fazit gezogen, das z. B. in die klinische Bewertung einfließt. Darüber hinaus werden CAPAs aufgeführt und begründet.Für Produkte der Klassen IIa, IIb und III wird ein PSUR erstellt, der – wie der PMS-Bericht – eine Zusammenfassung der Ergebnisse aus Marktdaten des jüngsten Erhebungszeitraums enthält. Auf Basis dieser Ergebnisse wird ein Fazit gezogen, das in die klinische Bewertung einfließt, und CAPAs werden aufgeführt und begründet. Zusätzlich zu den Inhalten des PMS-Berichts umfasst der PSUR die Ergebnisse der Nutzen-Risiko-Bewertung, die wichtigsten Erkenntnisse aus PMCF, die Verkaufszahlen des Produkts sowie weitere Informationen, z. B. zur Nutzungshäufigkeit.Ziel sowohl des PMS-Berichts als auch des PSUR ist es, Erkenntnisse über die Sicherheit und Leistung des Produkts auf dem Markt über den gesamten Lebenszyklus zu gewinnen, die für die weitere Produktentwicklung genutzt werden können und die jederzeit die Produktsicherheit gewährleisten. Der PMS-Bericht wird nach Bedarf erstellt und sollte kurz vor jeder Aktualisierung der klinischen Bewertung überarbeitet werden. Der PSUR wird regelmäßig (auch für Bestandsprodukte) gemäß der Risikoklasse des Produkts aktualisiert.
+SSCP – Summary of Safety and Clinical Performance (Zusammenfassung von Sicherheit und klinischer Leistung)
Der SSCP-Bericht muss für Produkte der Klasse III und für Implantate erstellt werden. Ziel des Kurzberichts ist es, das Produkt im Kontext seiner Verwendung darzustellen, Restrisiken und mögliche unerwünschte Wirkungen, Warnungen und Vorsichtsmaßnahmen zu erläutern und alternative Therapie- oder Diagnoseoptionen darzustellen. Der SSCP wird in der Gebrauchsanweisung oder Kennzeichnung referenziert und über EUDAMED der Öffentlichkeit zugänglich gemacht. Er muss so verfasst sein, dass er auch von Laien verstanden werden kann, sofern diese zur Zielgruppe gehören. Der SSCP wird jährlich aktualisiert.
Erfolgreiche Anpassung an die MDR: Unsere Whitepaper-Reihe für Ihre Medizinprodukte
Haben Sie bereits Ihre technische Dokumentation an die (EU) 2017/745 (MDR) angepasst? Falls nicht, wird es Zeit zu handeln und sicherzustellen, dass die Technische Dokumentation Ihrer Medizinprodukte den Anforderungen der MDR entspricht. Nur so können Sie die Sicherheit, Qualität und Wirksamkeit gewährleisten. In unserer Whitepaper-Reihe finden Sie wertvolle bewährte Verfahren zur Anpassung Ihrer bestehenden Produkte an die MDR. Starten Sie direkt mit Teil 1: "Umstellung Ihrer Bestandsprodukte" um den Marktzugang Ihrer Produkte in der EU zu erhalten!
Laden Sie sich jetzt unser Whitepaper kostenlos herunter
Laden Sie sich jetzt unser Whitepaper kostenlos herunter



