
Maßgeschneiderte Medizin - Sonderanfertigungen unter der Medical Device Regulation (MDR)
29.01.2024
Sie haben Fragen zum Beitrag oder möchten mehr über unsere Leistungen erfahren? Wir freuen uns auf Ihre Nachricht!Jetzt unverbindlich anfragen
Medizinprodukte unter der Medical Device Regulation (EU) 2017/745 (MDR) umfassen eine breite Palette an verschiedenen Produkten: von Einwegspritzen, die millionenfach hergestellt werden, bis hin zu Magnetresonanztomographie-Geräten, die mit ca. 2900 Geräten in Deutschland (Stand 2021) deutlich seltener vertreten sind. Doch es gibt auch Produkte, die wirklich nur einmalig hergestellt werden: Sonderanfertigungen.Doch was genau macht eine Sonderanfertigung aus und was muss dabei beachtet werden?Im Folgenden beschreiben wir, was Sonderanfertigungen gemäß MDR ausmacht und welche Implikationen dies für Hersteller hat, deren Produkte unter diese Definition fallen.
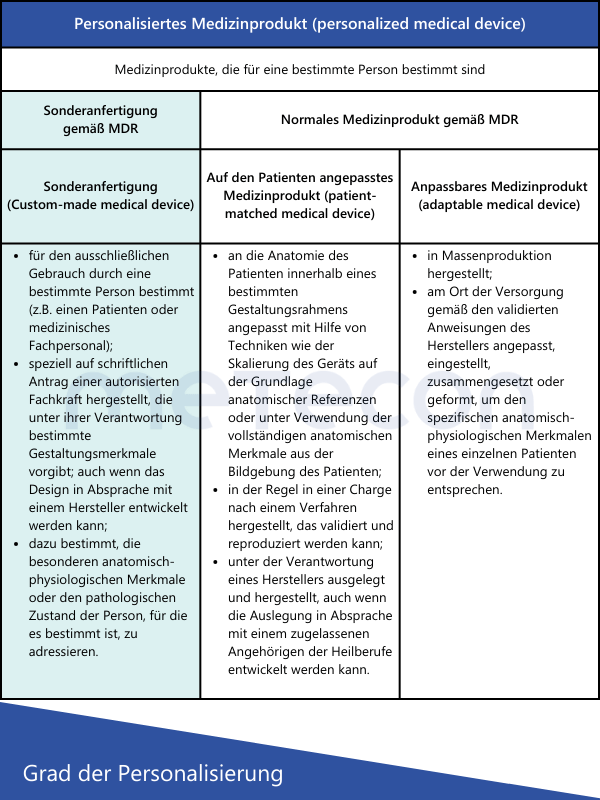
Aus den Definitionen ergeben sich folgende Schlussfolgerungen in Bezug auf Sonderanfertigungen:
Die MDR unterscheidet nur zwischen "normalen" Medizinprodukten und Sonderanfertigungen, daher gibt es auch nur für diese spezielle Anforderungen in der MDR. Eine weitere Unterteilung wie durch die MDCG und IMDRF ist daher zwar hilfreich, hat regulatorisch aber keine Auswirkungen auf einzelne Anforderungen.
Beste GrüßeLeon Weißenhorn
 Leon Weißenhorn
Leon Weißenhorn
Was sind Sonderanfertigungen?
Die MDR definiert Sonderanfertigungen in Artikel 2 Punkt 3 wie folgt:"Sonderanfertigung" bezeichnet ein Produkt, das speziell gemäß einer schriftlichen Verordnung einer aufgrund ihrer beruflichen Qualifikation nach den nationalen Rechtsvorschriften zur Ausstellung von Verordnungen berechtigten Person angefertigt wird, die eigenverantwortlich die genaue Auslegung und die Merkmale des Produkts festlegt, das nur für einen einzigen Patienten bestimmt ist, um ausschließlich dessen individuelle Zustand und dessen individuellen Bedürfnissen zu entsprechen.Serienmäßig hergestellte Produkte, die angepasst werden müssen, um den spezifischen Anforderungen eines berufsmäßigen Anwenders zu entsprechen, und Produkte, die gemäß den schriftlichen Verordnungen einer dazu berechtigten Person serienmäßig in industriellen Verfahren hergestellt werden, gelten jedoch nicht als SonderanfertigungenIn dieser Definition wird auch explizit genannt, welche Produktarten keine Sonderanfertigungen sind. Das International Medical Device Regulators Forum (IMDRF) hat dazu eigene Definitionen aufgestellt, die auch von der MDCG 2021-3 aufgegriffen werden. Zusätzlich zu dem Begriff "Sonderanfertigung" tauchen hier auch Begriffe wie "personalisiertes Medizinprodukt", "auf den Patienten angepasstes Medizinprodukt" und "anpassbares Medizinprodukt" auf. Beschäftigt man sich zum ersten Mal mit solchen Medizinprodukten, stellt sich die Frage: Sind diese Begriffe nur Synonyme oder worin liegen die Unterschiede?Die nachstehende Tabelle liefert eine kurze Gegenüberstellung der verschiedenen Definitionen nach IMDRF und was diese ausmachen.Aus den Definitionen ergeben sich folgende Schlussfolgerungen in Bezug auf Sonderanfertigungen:
- Alle Produkte, die serienmäßig hergestellt werden, auch wenn sie auf individuelle Anforderungen des Anwenders eingehen, sind keine Sonderanfertigungen.
- Das Produkt muss auf Basis einer schriftlichen Verordnung einer berechtigten Person angefordert werden, um als Sonderanfertigung gelten zu können.
- Diese Person muss die spezifische Auslegung des Produktes (z.B. Anzahl und Position von Schrauben, geometrischen Dimensionen) festlegen und ist auch dafür verantwortlich.
- Das Produkt darf nur für einen speziellen Patienten bestimmt sein, um als Sonderanfertigung zu gelten.
Die MDR unterscheidet nur zwischen "normalen" Medizinprodukten und Sonderanfertigungen, daher gibt es auch nur für diese spezielle Anforderungen in der MDR. Eine weitere Unterteilung wie durch die MDCG und IMDRF ist daher zwar hilfreich, hat regulatorisch aber keine Auswirkungen auf einzelne Anforderungen.
Worauf müssen Hersteller von Sonderanfertigungen achten?
Für Hersteller von Sonderanfertigungen fallen einige ansonsten notwendige Tätigkeiten weg, manche ändern sich und wieder andere kommen neu hinzu. Einige dieser Tätigkeiten werden im Folgenden näher erläutert.Anmerkung: Für Sonderanfertigungen der Klasse III oder implantierbare Sonderanfertigungen gelten weitere Besonderheiten. Auf diese wird in diesem Beitrag nicht eingegangen.Aspekte, die sich nicht ändern (keine vollständige Auflistung)
- Der Hersteller ist weiterhin verpflichtet die Grundlegenden Sicherheits- und Leistungsanforderungen einzuhalten (MDCG 2021-3, Punkt 8).
- Es wird ein Konformitätsbewertungsverfahren durchgeführt (MDCG 2021-3, Punkt 9).
- PMS, PMCF, Risikomanagement und eine klinische Bewertung müssen durchgeführt werden (MDCG 2021-3, Punkt 8).
- Es wird eine Person Responsible for Regulatory Compliance (PRRC) benötigt (MDR: Artikel 15).
Aspekte, die unterschiedlich betrachtet werden (keine vollständige Auflistung)
- Es wird keine EU-Konformitätserklärung erstellt. Somit wird auch kein CE-Kennzeichen auf dem Produkt angebracht. Die notwendige Konformitätsbewertung kann nur mittels des Verfahrens nach Anhang XIII durchgeführt werden (MDR: Artikel 10, Punkt 6, Artikel 20 Punkt 1, Artikel 21 Punkt 1).
- Die Zusammenarbeit mit einer Benannten Stelle im Rahmen der Konformitätsbewertung ist für Klasse IIa und IIb nicht notwendig (MDCG 2021-3, Punkt 9).
- Es muss keine Unique Device Identification (UDI) vergeben oder angebracht werden (MDR: Erwägungsgründe Punkt 42, Artikel 27 Punkt 1, 3, Artikel 29 Punkt 1,2,4).
- Die zuständigen Behörden können verlangen, dass der Hersteller ihnen eine Liste zur Verfügung stellt, die alle Sonderanfertigungen beinhaltet, die in dem jeweiligen Hoheitsgebiet in Verkehr gebracht wurden (MDR: Artikel 21, Punkt 2).
- Hersteller, Bevollmächtigter und Importeur müssen sich nicht in das in Artikel 30 genannte elektronische System (EUDAMED) eintragen (MDR: Artikel 30, Punkt 3; Artikel 31, Punkt 1).
- Die PRRC muss sich ebenfalls nicht in EUDAMED registrieren (MDCG 2021-3, Punkt 9).
- Es ist, anders als sonst üblich, bei Sonderanfertigungen möglich, die Qualifikation der PRRC über zwei Jahre Berufserfahrung in einem relevanten Bereich der Fertigung nachzuweisen (MDR: Artikel 15 Punkt 1).
- Auf dem Produkt und der Verpackung muss eine Aufschrift "Sonderanfertigung" vorhanden sein (MDR: Anhang I 23.2 und 23.3).
- Den Sonderanfertigungen muss eine Erklärung gem. Anhang XIII Abschnitt 1 MDR beigefügt sein (MDR: Artikel 21, Punkt 2). Der zugehörige Anwender oder Patient muss durch seinen Namen, ein Akronym oder einen numerischen Code identifizierbar sein (MDR: Artikel 21 Punkt 2).
- Es wird keine technische Dokumentation erstellt. Allerdings wird eine Dokumentation gem. Anhang XIII Abschnitt 2 gefordert (MDR: Artikel 10, Punkt 5).
Fazit
Wie auch bei den meisten anderen regulatorischen Fragestellungen müssen bei der Betrachtung von Sonderanfertigungen unter der MDR viele Details beachtet werden. Die Einordnung eines Medizinproduktes als Sonderanfertigung bringt einige Veränderungen für Hersteller mit sich - weder ein unüberwindbares Hindernis noch eine irrelevante Nebensächlichkeit.Benötigen Sie Unterstützung mit dem Konformitätsbewertungsverfahren für Ihre Sonderanfertigungen? Oder befassen Sie sich gerade mit einem anderen komplexen Thema aus den Bereichen Clinical Affairs oder Technische Dokumentation und suchen daher nach erfahrenen Fachleuten? Melden Sie sich gerne jederzeit bei uns. Gemeinsam finden wir die beste Lösung für Ihr konkretes Anliegen.Beste GrüßeLeon Weißenhorn
Unsere Blogbeiträge werden mit höchster Sorgfalt recherchiert und erstellt, sind jedoch lediglich Momentaufnahmen in der Regulatorik, und diese ist in stetem Wandel. Wir gewährleisten nicht, dass ältere Inhalte noch aktuell und aussagekräftig sind. Wenn Sie nicht sicher sind, ob der Beitrag, den Sie auf dieser Seite gelesen haben, noch dem aktuellen Stand der Regulierung entspricht, nehmen Sie bitte Kontakt zu uns auf: Wir ordnen Ihr Thema schnell in den aktuellen Kontext ein.
Regulatory Affairs & Technical Documentation



