
Leistungsbewertung: Wegweiser für die Erbringung der klinischen Leistung
29.08.2023
Sie haben Fragen zum Beitrag oder möchten mehr über unsere Leistungen erfahren? Wir freuen uns auf Ihre Nachricht!Jetzt unverbindlich anfragen
Der klinische Nachweis (clinical evidence) für In-vitro-Diagnostika (IVD) begründet sich auf den drei Säulen wissenschaftliche Validität (scientific validity), analytische Leistung (analytical performance) und klinische Leistung (clinical performance). Für die meisten Hersteller ist klar, wie sie die wissenschaftliche Validität und die analytische Leistung für ihr In-vitro-Diagnostikum nachweisen können. Der Nachweis der klinischen Leistung im Rahmen der Leistungsbewertung sorgt jedoch bei vielen Herstellern von IVD für Verunsicherung. Mit Blick auf die EU-Verordnung für In-vitro-Diagnostika (In vitro Diagnostic Medical Device Regulation (EU) 2017/746; IVDR) und das MedTech Europe E-Book Clinical Evidence Requirements under the EU In Vitro Diagnostics Regulation (IVDR) (Third Edition, February 2023) zeigen wir in diesem Beitrag auf, worauf es ankommt. Anhand von drei wesentlichen Fragestellungen veranschaulichen wir, wie Sie die klinische Leistung für ein bestimmtes Produkt nachweisen können.
Andere Quellen für klinische Leistungsdaten können sein:
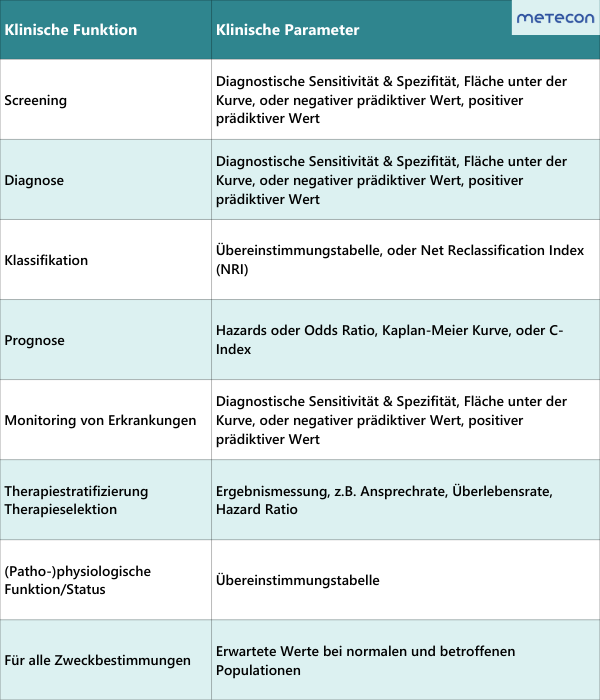 Abbildung 1: Klinische Leistung (Quelle: Clinical Evidence Requirements under the EU In Vitro Diagnostics Regulation (IVDR); Third Edition, February 2023; Seite 25)
Abbildung 1: Klinische Leistung (Quelle: Clinical Evidence Requirements under the EU In Vitro Diagnostics Regulation (IVDR); Third Edition, February 2023; Seite 25)
Klasse A < Klasse B < Klasse C < Klasse DDie Risikoklasse ist entscheidend für das Ausmaß des Nachweises der klinischen Leistung oder anders ausgedrückt: mit steigender Risikoklasse steigen auch die Anforderungen an die Daten zur klinischen Leistung. Je höher die Risikoklasse, desto eher brauchen Sie eine eigene klinische Leistungsstudie für Ihr Produkt. Umgekehrt räumt die MDCG 2022-2 sogar ein, dass für manche Klasse A Produkte gar keine Daten zur klinischen Leistung erforderlich sind:"The IVDR sets out that clinical performance may not be required for certain devices in Article 2 (39). For example, clinical performance data may not be expected for non-sterile specimen receptacles, microscopy glass slides, or some general reagents. In such cases and where due justification is given, a clinical performance report would not be expected. Nevertheless, the remaining aspects of the performance evaluation report including other elements of clinical evidence would still be required unless due justification is given."
Die Global Harmonization Task Force (GHTF) definiert etablierte In-vitro-Diagnostika (IVD) in ihrer Veröffentlichung GHTF/SG5/N7:2012 mit dem Titel "Clinical Evidence for IVD medical devices – Scientific Validity Determination and Performance Evaluation" (aus dem Jahr 2012!) als Tests , für die es klinische Leitlinien oder einen Konsens über die Anwendung gibt, sowie mehr als einen kommerziellen verfügbaren Test. Wenn es sich bei Ihrem IVD um einen etablierten Test handelt, schließt sich die Frage an, ob dieser auch standardisiert ist. Falls ja, ist ein internationaler Standard oder akzeptiertes Referenzmaterial für den Analyten erhältlich , mit dessen Hilfe der Test eingestellt werden muss (siehe auch GHTF/SG5/N7:2012 und MedTech E-Book Third Edition, February 2023). Die GHTF hebt hervor, dass etablierte und gleichzeitig standardisierte Tests unabhängig von der Methode oder vom Hersteller vergleichbare Ergebnisse für den Analyten liefern müssen.Ist Ihr In-Vitro-Diagnostikum (IVD) weder etabliert noch standardisiert, fällt Ihr Test in die Kategorie "neuartig". Neuartige Tests beinhalten gemäß GHTF/SG5/N7:2012:
Generell gilt: Ein Nachweis rein über die Äquivalenzroute sollte vermieden werden. Die Rückmeldungen und der Austausch mit Benannten Stellen haben uns gezeigt, dass Äquivalenz bei IVD nur sehr schwer nachzuweisen ist und folglich so gut wie nie akzeptiert wird.
Ein Beispiel: Wenn ein IVD zur Diagnose eines Eisenmangels bei Erwachsenen dient, müssen Proben von Erwachsenen verwenden werden, bei denen klar ist, ob ein Eisenmangel vorlag oder nicht. Die Verknüpfung der Proben mit einer klinischen Information ist elementar und unterscheidet letztlich analytische von klinischen Leistungsstudien. Auch für analytische Leistungsstudien kann Probenmaterial z. B. von Biobanken eingekauft werden, um die Genauigkeit (Accuracy) des eigenen In-vitro-Diagnostikums (IVD) im Vergleich mit einer Referenzmethode zu ermitteln. Hierbei handelt es sich um einen analytischen Methodenvergleich, bei dem bspw. das Bestimmtheitsmaß bestimmt wird.Bei einem Methodenvergleich im Rahmen des klinischen Leistungsnachweises, in denen das eigene IVD mit einer CE-markierten Referenzmethode verglichen wird, sollte am Ende der Studie z. B. das positive und negative Agreement (PNA) zwischen zwei In-vitro-Diagnostika (IVD) bestimmt werden. Nur mit dieser Bestimmung ist die Verknüpfung mit der klinischen Information entsprechen der Zweckbestimmung gegeben.
Beste Grüße
 Dr. Ulrike Kolbe
Dr. Ulrike Kolbe
 Dr. Sandra Reuter
Dr. Sandra Reuter
1 Wissenschaftliche Validität
Der Nachweis der wissenschaftlichen Validität bezeichnet "den Zusammenhang eines Analyten mit einem bestimmten klinischen oder physiologischen Zustand" (IVDR, Artikel 2 (38)). Dieser beruht in der Regel auf wissenschaftlicher, publizierter Literatur (peer-reviewed!) über den Analyten und den medizinischen Zustand oder die Krankheit. Untermauert werden sollte die Anwendung des Analyten und/oder der Technologie durch einvernehmliche Expertengutachten /-stellungnahmen oder Guidelines einschlägiger Fachorganisationen. Was, wenn die Literaturrecherche bei neuartigen Analyten/Markern/Technologien keine oder nur unzureichende Daten liefert? In diesen Fällen muss die wissenschaftliche Validität mithilfe eigener Studien zum Nachweis des Wirkprinzips (proof of concept studies) und/oder anhand der Ergebnisse klinischer Leistungsstudien (clinical performance studies) nachgewiesen werden.2 Analyseleistung
Die Analyseleistung bezeichnet "die Fähigkeit eines Produkts, einen bestimmten Analyten korrekt nachzuweisen oder zu messen" (IVDR, Artikel 2 (40)). Für den Nachweis der Analyseleistung gilt es, die Parameter gemäß IVDR (Anhang I, Abschnitt 9.1. a) nachzuweisen oder, bei Nichtanwendbarkeit, begründet wegzulassen. Um die Analyseleistung zu belegen, müssen eigene analytische Leistungsstudien durchgeführt werden. Die Durchführung der analytischen Leistungsstudien sollte sich, falls vorhanden, an gemeinsamen Spezifikationen und/oder an produktspezifischen Normen orientieren. Allgemeine, anerkannte Protokolle zur Bestimmung der analytischen Leistungsparameter liefern die entsprechenden CLSI-Guidelines.Achten Sie darauf, dass Sie nach den neuesten Versionen der Normen und Guidelines arbeiten. Die Benannten Stellen legen Wert darauf, dass Ihre Daten nach dem aktuellen Stand der Technik (State of the Art, SOTA) generiert und ausgewertet wurden.3 Klinische Leistung
Während das Vorgehen bis hierher klar ist und es wenig Spielraum für Interpretation gibt, gestaltet sich der Nachweis der klinischen Leistung etwas komplizierter, da mehrere Faktoren beachtet werden müssen. Die klinische Leistung bezeichnet "die Fähigkeit eines Produkts, Ergebnisse zu liefern, die mit einem bestimmten klinischen Zustand oder physiologischen oder pathologischen Vorgang oder Zustand bei einer bestimmten Zielbevölkerung und bestimmten vorgesehenen Anwendern korrelieren" (IVDR, Artikel 2 (41)). Auf den ersten Blick erscheint auch der Nachweis der klinischen Leistung klar definiert, da die IVDR, ebenso wie bei der Analyseleistung, nachzuweisende Parameter vorgibt (Anhang I. Abschnitt 9.1. b). Erneut gilt: bei Nichtanwendbarkeit können Parameter begründet weggelassen werden.Anders als bei der Analyseleistung ist es jedoch nicht zwingend erforderlich eigene klinische Leistungsstudien durchzuführen: "Auf die Durchführung klinischer Leistungsstudien gemäß Anhang XIII Teil A Abschnitt 2 kann nur dann verzichtet werden, wenn es ausreichende Gründe dafür gibt, auf andere Quellen klinischer Leistungsdaten zurückzugreifen" (IVDR, Artikel 56 (4)).Andere Quellen für klinische Leistungsdaten können sein:
- wissenschaftliche Literatur, die einem Peer-Review unterzogen wurde, und/oder
- Erfahrungen aus veröffentlichten, diagnostischen Routinetests.
3.1 Welche klinische Funktion erfüllt mein IVD?
Schauen Sie sich Ihre Zweckbestimmung genau an. Welche klinische Funktion (z. B. Diagnose/Hilfe zur Diagnose oder Monitoring) wird versprochen? Ist diese klinische Funktion mit einer konkreten Erkrankung verknüpft (z. B. Rheumatische Arthritis) oder lediglich mit einem (patho-)physiologischen Zustand (z. B. Eisenmangel)? Vergessen Sie dabei nicht, dass jegliche zusätzliche Behauptung in Ihren Marketingmaterialien und auf Ihrer Website die Zweckbestimmung erweitern. Die genaue Definition der klinischen Funktion Ihres IVD ist maßgeblich für die Auswahl der anwendbaren klinischen Leistungsparameter und für das Studiendesign zur Bestimmung dieser Parameter. Eine erste Orientierung, welche Parameter im Hinblick auf die klinische Funktion nachgewiesen werden sollten, erhalten Sie durch das MedTech Europe E-Book (Third Edition, February 2023). Grundsätzlich hängt es immer vom individuellen Produkt ab, welche Parameter sinnvoll sind. Für Rückfragen dazu stehen wir Ihnen gerne zur Verfügung.Abbildung 1: Klinische Leistung (Quelle: Clinical Evidence Requirements under the EU In Vitro Diagnostics Regulation (IVDR); Third Edition, February 2023; Seite 25)3.2 Welche Risikoklasse hat mein IVD?
Die Stärke und Robustheit des klinischen Nachweises sollen einem Risiko-basierten Ansatz folgen:Klasse A < Klasse B < Klasse C < Klasse DDie Risikoklasse ist entscheidend für das Ausmaß des Nachweises der klinischen Leistung oder anders ausgedrückt: mit steigender Risikoklasse steigen auch die Anforderungen an die Daten zur klinischen Leistung. Je höher die Risikoklasse, desto eher brauchen Sie eine eigene klinische Leistungsstudie für Ihr Produkt. Umgekehrt räumt die MDCG 2022-2 sogar ein, dass für manche Klasse A Produkte gar keine Daten zur klinischen Leistung erforderlich sind:"The IVDR sets out that clinical performance may not be required for certain devices in Article 2 (39). For example, clinical performance data may not be expected for non-sterile specimen receptacles, microscopy glass slides, or some general reagents. In such cases and where due justification is given, a clinical performance report would not be expected. Nevertheless, the remaining aspects of the performance evaluation report including other elements of clinical evidence would still be required unless due justification is given."
3.3 Ist mein IVD bereits etabliert am Markt?
Ein weiterer wichtiger Faktor, der Einfluss auf Art und Umfang des Nachweises der klinischen Leistung hat, ist die Neuartigkeit des In-vitro-Diagnostikums (IVD):- Ist ihr Produkt etabliert und/oder bereits unter der In-vitro-Diagnostic Device Directive (IVDD, abgelöst durch IVDR) am Markt zugelassen (= legacy device; Legacy-Produkt)?
- Soll das Produkt erstmalig in Verkehr gebracht werden?
- Ist die Technologie hinter dem Produkt neuartig?
- Gibt es bereits viele vergleichbare Produkte am Markt?
Die Global Harmonization Task Force (GHTF) definiert etablierte In-vitro-Diagnostika (IVD) in ihrer Veröffentlichung GHTF/SG5/N7:2012 mit dem Titel "Clinical Evidence for IVD medical devices – Scientific Validity Determination and Performance Evaluation" (aus dem Jahr 2012!) als Tests , für die es klinische Leitlinien oder einen Konsens über die Anwendung gibt, sowie mehr als einen kommerziellen verfügbaren Test. Wenn es sich bei Ihrem IVD um einen etablierten Test handelt, schließt sich die Frage an, ob dieser auch standardisiert ist. Falls ja, ist ein internationaler Standard oder akzeptiertes Referenzmaterial für den Analyten erhältlich , mit dessen Hilfe der Test eingestellt werden muss (siehe auch GHTF/SG5/N7:2012 und MedTech E-Book Third Edition, February 2023). Die GHTF hebt hervor, dass etablierte und gleichzeitig standardisierte Tests unabhängig von der Methode oder vom Hersteller vergleichbare Ergebnisse für den Analyten liefern müssen.Ist Ihr In-Vitro-Diagnostikum (IVD) weder etabliert noch standardisiert, fällt Ihr Test in die Kategorie "neuartig". Neuartige Tests beinhalten gemäß GHTF/SG5/N7:2012:
- einen neuen Analyten,
- eine neue Technologie,
- eine neue Anwendung einer etablierten Technologie,
- eine neue Zielgruppe oder
- eine neue Zweckbestimmung.
4 Nachweis der klinischen Leistung: neuartige Produkte versus Legacy-Produkte
Ist Ihr Produkt neuartig, müssen Sie eine eigene prospektive und/oder retrospektive klinische Leistungsstudien durchführen, um die klinische Leistung oder zumindest den "neuartigen" Teil der klinischen Leistung (z .B. Indikationserweiterung) belegen zu können. Auch für Produkte der höchsten Risikoklassen (Klasse C und D) sollten klinische Leistungsstudien immer in Betracht gezogen werden. Die eigene klinische Leistungsstudie trägt als Quelle für die klinische Leistung die höchste Aussagekraft, da das Studiendesign genau auf die zu bestimmenden Parameter angepasst werden kann.Ist Ihr Produkt bereits am Markt, können auch Daten und Ergebnisse zur klinischen Leistung des Produktes aus peer-reviewed Literatur herangezogen werden.Achtung: Hier muss genau analysiert werden, ob das Studiendesign, die Studiendurchführung, die Zahl der eingeschlossenen Probanden, die Methodik, die Datenanalyse etc. geeignet ist, bzw. die Nutzung der Daten für den Nachweis der klinischen Leistung des Produktes erlaubt. Diese publizierten Studien werden meist unabhängig vom Hersteller durchgeführt und veröffentlicht. Aus diesem Grund entsprechen sie nicht immer dem optimalen Studiendesign, das der Hersteller zur Demonstration der klinischen Leistung benötigt. Wenngleich diese Studien nicht immer alle Parameter abdecken, die Sie für Ihr IVD nachweisen müssen, können diese zur Unterstützung und/oder Untermauerung der klinischen Leistung herangezogen werden.Für Produkte, die bereits einige Zeit am Markt sind bzw. bereits unter IVDD auf dem Markt waren (= legacy device; Legacy-Produkt), ist es durchaus üblich, eigene Studiendaten, die unter der IVDD generiert wurden, mit Daten aus peer-reviewed Literatur zu untermauern. So muss keine neue klinische Leistungsstudie nach IVDR durchgeführt werden. Voraussetzung zur Verwendung solch "anderer" Studiendaten ist, dass das Studiendesign, die Durchführung und die Zahl der Proben noch dem Stand der Technik (SOTA) entsprechen und somit den Qualitätsansprüchen genügen.Es kommt auf die Datenlage an, ob Sie selbst klinische Leistungsstudien durchführen oder sich auf eine Mischung aus alten eigenen Daten und publizierter, peer-reviewed Literatur über Ihr Legacy-Produkt berufen. Starten Sie mit einer Gap-Analyse Ihrer vorhanden klinischen Studiendaten. Wenn Sie Lücken finden, können Sie eine gut strukturierte und umfassende Literatursuche mit wohlüberlegten Suchbegriffen aufsetzen. Dies hilft Ihnen zu entscheiden, ob Sie Ihre Lücken über die publizierte Literatur schließen können, bzw. zeigt Ihnen auf, welche Aspekte von klinischer Relevanz (z. B. Indikationen, Zielgruppen) noch nicht ausreichend abgedeckt sind. Für nicht abgedeckte Aspekte müssten Sie eigene Daten generieren.5 Nachweis der klinischen Leistung per Methodenvergleich
Wenn es sich bei Ihrem In-vitro-Diagnostikum (IVD) um einen etablierten Test handelt, müssen Sie nicht unbedingt eine prospektive oder retrospektive Leistungsstudie durchführen. Sie können zum Nachweis der klinischen Leistung auch einen Methodenvergleich zwischen Ihrem Produkt und einem CE-gekennzeichneten Referenzprodukt vornehmen. Dies gilt sowohl für hohe als auch niedrige Risikoklassen. Wichtig ist, dass die Referenzmethode dem Stand der Technik (SOTA) entspricht und die klinische Leistung der Referenzmethode inklusive zutreffender klinischer Leistungsparameter in der Leistungsbewertung dargestellt wird. Die klinische Leistung des Referenzprodukts sollte dementsprechend bekannt und veröffentlicht sein (z. B. durch die Gebrauchsanweisung; IFU, Instructions for use).5.1 Vorsicht vor Äquivalenzbetrachtungen bei In-vitro-Diagnostika!
Wenn Ihr IVD nicht nur etabliert, sondern auch standardisiert ist, kann es laut den Autoren des MedTech E-Book (Third Edition, February 2023) ausreichen, die klinischen Leistungsparameter über die Literatur oder mittels veröffentlichter Daten zu einem standardisierten Referenzprodukt nachzuweisen. Dies gälte, insofern die Bestimmung der analytischen Leistung Ihres In-vitro-Diagnostikums (IVD) unter Verwendung von standardisiertem Produkt- und Referenzmaterial erfolgt ist.Unsere Empfehlung: Führen Sie stets einen Methodenvergleich mit einem CE-gekennzeichneten Referenzprodukt durch. Uns ist bisher kein Fall bekannt, bei dem der Nachweis der klinischen Leistung ausschließlich über die Literatur zu einem Referenzprodukt erfolgt ist UND dies von Benannten Stellen akzeptiert wurde.Generell gilt: Ein Nachweis rein über die Äquivalenzroute sollte vermieden werden. Die Rückmeldungen und der Austausch mit Benannten Stellen haben uns gezeigt, dass Äquivalenz bei IVD nur sehr schwer nachzuweisen ist und folglich so gut wie nie akzeptiert wird.
5.2 Hinweis für die Durchführung von Methodenvergleichen für den Nachweis der klinischen Leistung
Bei retrospektiven, klinischen Leistungsstudien ist es sehr wichtig, dass die von Ihnen verwendeten Proben die Zielpopulation, wie in der Zweckbestimmung angegeben, widerspiegeln.Ein Beispiel: Wenn ein IVD zur Diagnose eines Eisenmangels bei Erwachsenen dient, müssen Proben von Erwachsenen verwenden werden, bei denen klar ist, ob ein Eisenmangel vorlag oder nicht. Die Verknüpfung der Proben mit einer klinischen Information ist elementar und unterscheidet letztlich analytische von klinischen Leistungsstudien. Auch für analytische Leistungsstudien kann Probenmaterial z. B. von Biobanken eingekauft werden, um die Genauigkeit (Accuracy) des eigenen In-vitro-Diagnostikums (IVD) im Vergleich mit einer Referenzmethode zu ermitteln. Hierbei handelt es sich um einen analytischen Methodenvergleich, bei dem bspw. das Bestimmtheitsmaß bestimmt wird.Bei einem Methodenvergleich im Rahmen des klinischen Leistungsnachweises, in denen das eigene IVD mit einer CE-markierten Referenzmethode verglichen wird, sollte am Ende der Studie z. B. das positive und negative Agreement (PNA) zwischen zwei In-vitro-Diagnostika (IVD) bestimmt werden. Nur mit dieser Bestimmung ist die Verknüpfung mit der klinischen Information entsprechen der Zweckbestimmung gegeben.
Fazit
Um zu ermitteln, wie Sie den klinischen Nachweis für Ihr In-vitro-Diagnostikum (IVD) erbringen können, schauen Sie sich zunächst die klinische Funktion in der Zweckbestimmung genau an, um herauszufinden, welche klinischen Leistungsparameter für Ihr IVD anwendbar sind. Anhand der Risikoklasse und der Etablierung am Markt sowie einer eventuellen Standardisierung bestimmen Sie, ob ein direkter Nachweis (Nachweis der klinischen Leistungsparameter mit Daten zu Ihrem eigenen Produkt) der klinischen Leistung erforderlich ist oder ob ein indirekter Nachweis ausreicht (z. B. Methodenvergleich und Darstellung der zutreffenden klinischen Leistungsparameter der CE-markierten Referenzmethode). Unsere IVD-Expert*innen unterstützen Sie gerne bei allen Fragen rund um die Leistungsbewertung (performance evaluation ). Dabei kann sich unsere Unterstützung auf eine Strategie-Entwicklung beschränken oder auch die konkrete Umsetzung umfassen. Wir freuen uns auch über Fragen abseits von Clinical Affairs – vereinbaren Sie jetzt Ihr kostenloses Erstgespräch!Beste Grüße
Unsere Blogbeiträge werden mit höchster Sorgfalt recherchiert und erstellt, sind jedoch lediglich Momentaufnahmen in der Regulatorik, und diese ist in stetem Wandel. Wir gewährleisten nicht, dass ältere Inhalte noch aktuell und aussagekräftig sind. Wenn Sie nicht sicher sind, ob der Beitrag, den Sie auf dieser Seite gelesen haben, noch dem aktuellen Stand der Regulierung entspricht, nehmen Sie bitte Kontakt zu uns auf: Wir ordnen Ihr Thema schnell in den aktuellen Kontext ein.
Expert
Regulatory Affairs & Clinical Affairs
Team Lead IVD
Regulatory Affairs & Technical Documentation



