
Therapiebegleitende Diagnostika: Daran sollten Sie bei CDx denken!
22.11.2022
Sie haben Fragen zum Beitrag oder möchten mehr über unsere Leistungen erfahren? Wir freuen uns auf Ihre Nachricht!Jetzt unverbindlich anfragen
Die mittlerweile in Kraft getretene IVDR bringt viele Neuerungen mit, auf die sich In-vitro-Diagnostika-Hersteller einstellen müssen. Dazu gehört, dass in der IVDR erstmalig therapiebegleitende Diagnostika (Companion Diagnostics, CDx) definiert werden. Bislang wurden CDx wie "gewöhnliche" IVDs behandelt und unterlagen keinen speziellen Anforderungen. Die neue Definition bringt dagegen einige regulatorische Besonderheiten mit sich. Im folgenden Beitrag erfahren Sie, was das für Sie als Hersteller im Einzelnen bedeutet und worauf zu achten ist.
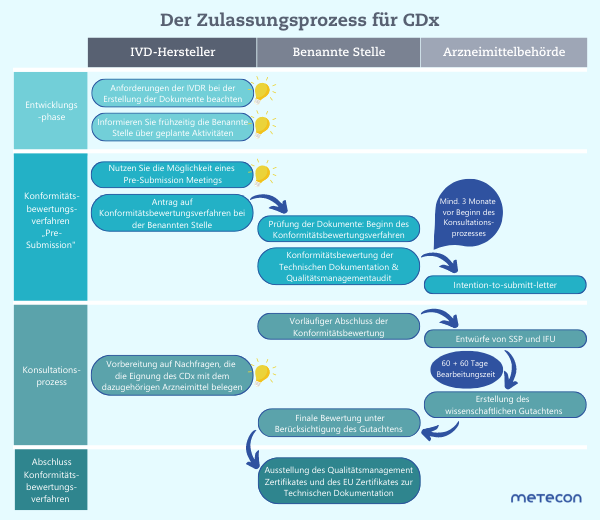
Durch dieses zusätzliche Verfahren verlängert sich der Zulassungsprozess um 6 bis 8 Monate. Davon ausgehend, dass das Konformitätsbewertungsverfahren für ein "normales" IVD bereits 12 Monate betragen kann, liegt die Gesamtdauer für ein CDx bei ca. 18 bis 20 Monaten. Eine regulatorische Besonderheit in Europa: Mit den Benannten Stellen und der Arzneimittelbehörde sind zwei unterschiedliche Behörden in diesen Prozess involviert, die wiederum unterschiedlichen Rechtsrahmen unterliegen. In den USA hingegen wird der gesamte Prozess einzig von der FDA durchgeführt.
Die Arzneimittelbehörde kann trotz vorliegender Entwürfe von SSP und IFU die Eignung in Frage stellen. Um bei Nachfragen die Eignung belegen zu können, ist es sinnvoll, diese Kernfrage in Ihrer Dokumentation klar und deutlich auszuarbeiten. Die Arzneimittelbehörde hat insgesamt 120 Tage Zeit für die Erstellung des wissenschaftlichen Gutachtens – das sollten Sie bei der Bearbeitung von Nachfragen im Blick behalten.
Für größere Ansicht Grafik anklicken
Beste Grüße
 Dr. Sandra Neidhöfer
Dr. Sandra Neidhöfer
Was sind therapiebegleitende Diagnostika (CDx)?
Bei einem therapiebegleitenden Diagnostikum handelt es sich laut Definition (IVDR) um ein Produkt, das für die sichere und wirksame Verwendung eines dazugehörigen Arzneimittels wesentlich ist, indem es Patient*innen vor und/oder während der Behandlung anhand von Biomarkern sortiert. Die identifizierten Patient*innen profitieren entweder von dem Arzneimittel (positive Biomarker) oder haben ein erhöhtes Risiko (negative Biomarker), dass eine schwerwiegende unerwünschte Reaktion als Folge der Behandlung mit dem Arzneimittel entsteht.Nicht zu den therapiebegleitenden Diagnostika werden Produkte gezählt, die zur Überwachung der Behandlung mit einem Arzneimittel eingesetzt werden. Ebenso gelten die sogenannten ergänzenden Diagnostika nicht als CDx, wobei die Abgrenzung nicht immer ganz einfach ist. Bei dem Begriff "complementary diagnostics" (ergänzende Diagnostika) handelt es sich um keinen europäischen Begriff und er ist regulatorisch nicht definiert. Wichtigstes Merkmal für ein CDx ist, dass es für die sichere Verwendung des Arzneimittels essenziell ist, während ergänzende Diagnostika lediglich eine Entscheidungshilfe darstellen. In den USA hingegen bindet die FDA (Food and Drug Administration) in ihrer Definition von "Companion Diagnostics" sowohl ergänzende als auch überwachende Diagnostika ein und erhebt die gleichen Anforderungen an diese Produkte. Unter der IVDR gibt es für therapiebegleitende Diagnostika einige Besonderheiten, während überwachende und ergänzende Diagnostika wie "gewöhnliche" IVDs behandelt werden. Im Rahmen der Zulassungen zählt dies zu einer der regulatorischen Hürden, mit denen CDx-Hersteller konfrontiert sind.Das hat sich mit der IVDR geändert
Unter der IVDD waren die therapiebegleitenden Diagnostika der Gruppe "Sonstige" zugeteilt und nicht näher beschrieben. Unter der IVDR fallen diese Produkte unter die Risikoklasse C (IVDR Anhang VIII, 2.3f), und müssen jeweils einzeln das Konformitätsbewertungsverfahren durchlaufen (IVDR, Artikel 48, 7).Die wichtigste Neuerung stellt der Einbezug der Arzneimittelbehörde im Konformitätsbewertungsverfahren dar. Im sogenannten Konsultationsprozess holt sich die Benannte Stelle ein Gutachten ein: entweder bei der für die Zulassung zuständigen Arzneimittelbehörde oder bei der EMA (Europäische Arzneimittel-Agentur) im Falle von Neuzulassungen. Nur unter Berücksichtigung dieses Gutachtens darf die Benannte Stelle eine finale Entscheidung treffen und bei positivem Ergebnis das Zertifikat für Qualitätsmanagement und das EU-Zertifikat zur Technischen Dokumentation ausstellen.Durch dieses zusätzliche Verfahren verlängert sich der Zulassungsprozess um 6 bis 8 Monate. Davon ausgehend, dass das Konformitätsbewertungsverfahren für ein "normales" IVD bereits 12 Monate betragen kann, liegt die Gesamtdauer für ein CDx bei ca. 18 bis 20 Monaten. Eine regulatorische Besonderheit in Europa: Mit den Benannten Stellen und der Arzneimittelbehörde sind zwei unterschiedliche Behörden in diesen Prozess involviert, die wiederum unterschiedlichen Rechtsrahmen unterliegen. In den USA hingegen wird der gesamte Prozess einzig von der FDA durchgeführt.
Was ist für den Konsultationsprozess wichtig?
Eine gute Kommunikation und gutes Timing sind das A und O. Sprechen Sie frühzeitig, vor Beginn des Konformitätsbewertungsverfahrens, mit Ihrer benannten Stelle und planen Sie zusammen die nächsten Schritte. Die EMA hat einen hilfreichen Leitfaden für den Konsultationsprozess veröffentlicht: Darin wird für jede Phase des Konsultationsprozesses eine Empfehlung ausgesprochen. Nutzen Sie zudem die von der EMA zur Verfügung gestellten Vorlagen. Das spart Zeit für Nachfragen und vermeidet Lücken im Informationsfluss.Dass eine gute zeitliche Taktung zwischen Benannter Stelle und Arzneimittelbehörde wichtig ist, zeigt sich bereits vor der Einreichung: Die Benannte Stelle informiert mindestens 3 Monate vor Beginn des Konsultationsprozesses die Arzneimittelbehörde in einem "intention-to-submit-letter". In dieser frühen Phase kann zusätzlich ein "Pre-Submission Meeting" zwischen Arzneimittelbehörde, Benannter Stelle, IVD-Hersteller und gegebenenfalls auch Arzneimittelhersteller stattfinden, um potenzielle Hürden zu identifizieren. Ergreifen Sie wenn möglich diese Chance!Die Konsultation beginnt erst nach der Konformitätsbewertung der Technischen Dokumentation durch die Benannte Stelle und basiert auf den Entwürfen für die Summary of Safety and Performance (SSP) und für die Gebrauchsanweisung (IFU). Es ist unerlässlich, dass beide Dokumente zuerst durch die Benannte Stelle geprüft und gegebenenfalls überarbeitet werden. Die Kernaufgabe der Arzneimittelbehörde im Konsultationsprozess ist es zu prüfen, ob sich das CDx für seine Verwendung mit dem entsprechenden Arzneimittel eignet.Die Arzneimittelbehörde kann trotz vorliegender Entwürfe von SSP und IFU die Eignung in Frage stellen. Um bei Nachfragen die Eignung belegen zu können, ist es sinnvoll, diese Kernfrage in Ihrer Dokumentation klar und deutlich auszuarbeiten. Die Arzneimittelbehörde hat insgesamt 120 Tage Zeit für die Erstellung des wissenschaftlichen Gutachtens – das sollten Sie bei der Bearbeitung von Nachfragen im Blick behalten.
Für größere Ansicht Grafik anklicken
Die Unterschiede in der Zulassung von ko-entwickeltem, Follow-on und Legat-CDx
Therapiebegleitende Diagnostika lassen sich typischerweise in drei Szenarien unterteilen: ko-entwickeltes CDx, Follow-on CDx und Legat-CDx.Ein ko-entwickeltes CDx wird zusammen mit dem dazugehörigen Arzneimittel entwickelt. Hier ist die frühzeitige Interaktion zwischen Benannter Stelle und Arzneimittelbehörde gefragt, um die Arzneimittel- und CDx- Zulassung bestmöglich aufeinander abzustimmen. Auch wenn es keine gesetzliche Vorgabe gibt, dass die Freigabe des Arzneimittels und die Zertifizierung des CDx gleichzeitig erfolgen müssen, ist dies sinnvoll. Die aktive Anwendung an Patient*innen kann erst beginnen, wenn sowohl das CDx als auch das dazugehörige Arzneimittel auf dem Markt zugelassen sind. Im Idealfall verwenden Sie für die klinische Studie ein bereits marktreifes CDx. Häufig ist dies allerdings nicht möglich, so dass ein klinischer Studien-Assay zum Einsatz kommt. Für diesen Fall benötigen Sie eine Konkordanz-Studie, um die Vergleichbarkeit des Produkts mit dem klinischen Studien-Assay zu belegen. Die klinische Leistung darf hierbei die sichere und effiziente Verwendung des Arzneimittels nicht beeinflussen.Ein Follow-on CDx weist den gleichen Biomarker nach wie ein zu einem Arzneimittel ko-entwickeltes CDx. Die Entwicklung erfolgt allerdings unabhängig und zeitlich versetzt von der Arzneimittel-Entwicklung. Stellen Sie sicher, dass das Follow-on CDx dem ko-entwickelten CDx in der sicheren und effizienten Verwendung des Arzneimittels in nichts nachsteht, und belegen Sie dies mit vergleichbaren analytischen und klinischen Leistungen.An dritter Stelle sind die Legat-CDx zu nennen. Diese Produkte wurden bereits unter der IVDD auf den Markt gebracht und sind seit Inkrafttreten der IVDR als CDx zu klassifizieren. Je nachdem, wie das CDx entwickelt wurde, kann es entweder einem ko-entwickelten CDx oder einem Follow-on CDx zugeordnet werden. Denken Sie daran: Signifikante Änderungen an Ihrem Legat-CDx sind unzulässig. Die Deadline für Produkte, bei denen das Konformitätsbewertungsverfahren bisher keine Mitwirkung der Benannten Stelle erfordert, ist der 26. Mai 2026. Nutzen Sie die Zeit, um die Anforderungen der IVDR an Ihr Produkt schnellstmöglich umzusetzen!Fazit – Was ist mit Änderungen nach dem Inverkehrbringen?
Bei Änderungen an einem therapiebegleitenden Diagnostikum nach dem Inverkehrbringen (unter IVDR) ist Vorsicht geboten. Wirken sich die Änderungen auf die Leistung, die bestimmungsgemäße Verwendung und/oder die Eignung des Produkts in Verbindung mit dem betreffenden Arzneimittel aus, muss die Benannte Stelle – zur Bewertung dieser Änderung – informiert werden. Entscheidet die Benannte Stelle, dass ein Nachtrag zu der EU-Bescheinigung über die Bewertung der Technischen Dokumentation notwendig ist, holt sie im Rahmen einer Follow-up-Konsultation ein Gutachten der Arzneimittelbehörde ein. Nach Eingang eines formalen Antrags, einschließlich der relevanten Dokumente, hat die Arzneimittelbehörde 30 Tage Zeit, um ein Gutachten zu erstellen. Sofern sich die Benannte Stelle für das Erfordernis einer neuen Konformitätsbewertung entscheidet, startet das Konsultationsverfahren von Neuem. Überlegen Sie daher gut, welche Änderungen Sie an Ihrem CDx (Companion Diagnostic) vornehmen, und informieren Sie Ihre Benannte Stelle im Vorfeld über geplante Aktivitäten!Wir unterstützen Sie gerne in allen Fragen rund um Ihr CDx. Und nicht nur das – wir sind Experten auf dem Gebiet der Regulatory Affairs und bieten Ihnen kompetente Beratung mit individuellen Lösungen an. Unsere Erstgespräche sind unverbindlich und kostenfrei, so dass Sie kein Risiko eingehen. Wir freuen uns auf Sie!Beste Grüße
Unsere Blogbeiträge werden mit höchster Sorgfalt recherchiert und erstellt, sind jedoch lediglich Momentaufnahmen in der Regulatorik, und diese ist in stetem Wandel. Wir gewährleisten nicht, dass ältere Inhalte noch aktuell und aussagekräftig sind. Wenn Sie nicht sicher sind, ob der Beitrag, den Sie auf dieser Seite gelesen haben, noch dem aktuellen Stand der Regulierung entspricht, nehmen Sie bitte Kontakt zu uns auf: Wir ordnen Ihr Thema schnell in den aktuellen Kontext ein.
IVD Expert
Regulatory Affairs



