
Schweiz: Neue Regulierung der In-vitro-Diagnostika durch IvDV
07.02.2023
Sie haben Fragen zum Beitrag oder möchten mehr über unsere Leistungen erfahren? Wir freuen uns auf Ihre Nachricht!Jetzt unverbindlich anfragen
Das Mutual Recognition Agreement (MRA) zwischen der Schweiz und der EU hat in der Vergangenheit das grenzüberschreitende Handeln mit Medizinprodukten/In-Vitro-Diagnostika erleichtert. Mit Inkrafttreten der EU-Verordnung 2017/746 über In-vitro-Diagnostika (IVDR) im Mai 2022 wurde eine neue Vereinbarung notwendig. Das Abkommen über die gegenseitige Anerkennung von Konformitätsbewertungen wurde jedoch nicht mehr aktualisiert.Die Schweiz ist seitdem EU-Drittstaat, und ihr Markt ist nicht mehr ungehindert zugänglich. Ein hindernisfreier gegenseitiger Marktzugang und eine gemeinsame Marktüberwachung sind nicht mehr gewährleistet. Für das Inverkehrbringen von IVDs benötigen EU-Hersteller einen Schweizer Bevollmächtigten und Importeur. Diese Anforderung ergibt sich aus der Schweizer Medizinprodukteverordnung (MepV; SR 812.213).Medizinprodukte, die in der Schweiz in Verkehr gebracht oder in Betrieb genommen werden, müssen die Anforderungen der MepV bzw. der Verordnung über In-vitro-Diagnostika (IvDV, SR 812.219) erfüllen. In diesem Beitrag erfahren sie mehr über die Regulierung der In-vitro-Diagnostika im Schweizer Markt.
Quelle: https://www.swiss-medtech.ch/en/news/overview-ivdo-dates-and-deadlines
In der Schweiz ansässige Importeure und Händler müssen das folgende Formular bei der Swissmedic einreichen:
In der EU/EWR ansässige Hersteller von IVDs müssen Ihre Produkte nicht separat bei der Swissmedic melden, wenn diese in der Schweiz in Verkehr gebracht werden. Vorausgesetzt die Produkte sind IVDR-konform (CE) und es wurde eine Person mit Sitz in der Schweiz als CH-Rep eingetragen!Seit dem 27. Mai 2022 werden keine IVD gemäß IVDD (98/79/EG) mehr auf dem Schweizer Markt neu zugelassen. Noch auf dem Markt befindliche IVDs (Legacy Devices) dürfen keine signifikanten Änderungen erfahren!
Beste Grüße
 Dunja Schildge-Reichman
Dunja Schildge-Reichman
Die neue IvDV gilt seit 26. Mai 2022
Am 4. Mai 2022 verabschiedete der Schweizer Bundesrat die neue IvDV sowie den Änderungserlass der Verordnung über klinische Versuche mit Medizinprodukten (KlinV-Mep). Sie bilden die letzte Etappe der Anpassung des Schweizer Medizinprodukterechts mit folgenden Zielen:- Verbesserung des Patientenschutzes durch strengere Vorgaben für die Konformitätsbewertung,
- Überwachung nach dem Inverkehrbringen und
- Angleichung an die neuen Vorschriften der Europäischen Union.
Die Situation in der EU
Die Umsetzung der IVDR konnte wegen der Herausforderungen der Covid-19-Pandemie, der begrenzten Kapazitäten der benannten Stellen sowie der Komplexität der Verordnung selbst bis zum Geltungsbeginn am 26. Mai 2022 nicht erfolgen. Deshalb erließ die EU am 25. Januar 2022 die Verordnung (EU) 2022/112 zur Änderung der Verordnung (EU) 2017/746 hinsichtlich der Übergangsbestimmungen für bestimmte In-vitro-Diagnostika und des späteren Geltungsbeginns der Bedingungen für hausinterne Produkte". Mit diesen nach Risikoklassen abgestuften und längstens bis im Jahr 2027 dauernden Übergangsfristen, beabsichtigt die EU eine drohende Versorgungslücke zu verhindern.Übergangsbestimmungen in der Schweiz
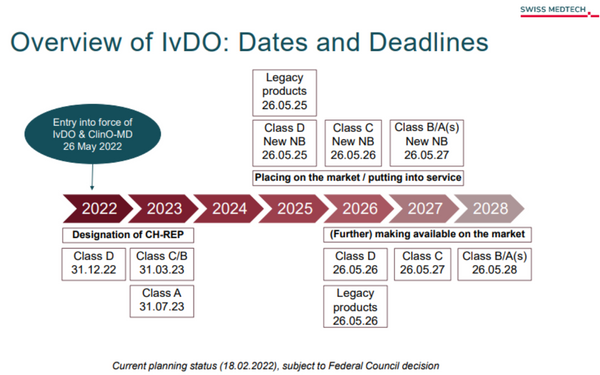
Diese neuen Übergangsfristen der EU sind auch in der IvDV entsprechend berücksichtigt. Die IvDV sieht ergänzend verschiedene Übergangsfristen und Maßnahmen vor, um die Versorgung der Schweiz mit sicheren IVDs aus der EU weiterhin zu gewährleisten. Folgende Maßnahmen sind geplant:1. EU-Konformitätsbescheinigungen werden anerkannt!2. Registrierung der Wirtschaftsakteure (Hersteller, Importeure und Bevollmächtigte): Die Registrierung erfolgt über die Swissmedic. Hier erhalten Akteure eine einmalige Registrierungsnummer, die Swiss Single Registration Number – CHRN.3. Die Meldung von schwerwiegenden Vorkommnissen und Sicherheitsberichten erfolgt ebenfalls bei der Swissmedic. Diese Meldung erfolgt durch den designierten CH-REP für die in der EU-ansässigen Hersteller von IVDs.4. Das Etablieren eines Schweizer Bevollmächtigten (CH-REP) für ausländische Hersteller soll dazu beigetragen, dass die Swissmedic die Marktüberwachung trotz Ausschluss aus dem Netzwerk der EU-Behörden aufrechterhalten kann. Hierfür gelten längere Übergangsfristen (siehe Abbildung).5. Erleichterung der Kennzeichnungspflicht: Die IvDV ermöglicht für alle Produkte, die durch Fachpersonen gehandhabt werden, eine alternative CH-REP-Angabe auf einem dem IVD beiliegenden Dokument (bspw. einem Lieferschein). Das gilt nicht für Produkte, die zur Eigenanwendung bestimmt sind und gemäß neuem Recht in Verkehr gebracht werden. Fristverlängerung bis 31. März 2025.Überblick über die IvDV: Termine und Fristen:Quelle: https://www.swiss-medtech.ch/en/news/overview-ivdo-dates-and-deadlines
Zulassung/Meldung von In-vitro-Diagnostika gemäß IvDV in der Schweiz
In der Schweiz ansässige Hersteller von IVDs müssen die Swissmedic informieren, sobald sie ihre Produkte initial auf dem Schweizer Markt in Verkehr bringen wollen.I. IVDs der Klassen D, C, B und A steril müssen individuell gemeldet werden. Hierfür stellt die Swissmedic zwei Formblätter zur Verfügung:- Formblatt für die Anzeige der Hersteller gem. Art.90 Abs.1 IvDV,
- Formblatt für Zertifizierungsdaten von In-vitro-Diagnostika Art. 90 Abs. 1 IvDV (IVDR Annex IX-XI).
- die Bescheinigung zu den durchgeführten Verfahren der Konformitätsbewertung (EG-Zertifikate), und
- die Gebrauchsanweisung, sowie für Produkte zur Eigenanwendung und für patientennahe Tests (POCT) – zusätzlich das Layout der äußeren Verpackung.
- Formblatt für die Anzeige der Hersteller gem. Art. 90 Abs.1 IvDV.
In der Schweiz ansässige Importeure und Händler müssen das folgende Formular bei der Swissmedic einreichen:
- Formblatt Meldung nach Art. 46 Abs.4 und Art. 47 Abs. 4 IvDV umgepackten/umgekennzeichneten In-vitro-Diagnostika.
- Selbst entwickelte, mit eigenen (nicht CE-markierten) Reagenzien durchgeführte, medizinisch-analytische Testverfahren;
- Auf Standardverfahren oder publizierten Verfahren beruhende, medizinisch-analytische Testverfahren, die mit eigenen (nicht CE-markierten) Reagenzien durchgeführt werden;
- Erworbene, aber nicht für die medizinische Anwendung vorgesehene Testverfahren (z. B. "Research Use Only/RUO"-Verfahren), die von der Gesundheitseinrichtung für die medizinisch-analytische Anwendung (weiter)entwickelt wurden;
- Selbst hergestellte IVD-Instrumente;
- Selbst entwickelte IVD-Software.
In der EU/EWR ansässige Hersteller von IVDs müssen Ihre Produkte nicht separat bei der Swissmedic melden, wenn diese in der Schweiz in Verkehr gebracht werden. Vorausgesetzt die Produkte sind IVDR-konform (CE) und es wurde eine Person mit Sitz in der Schweiz als CH-Rep eingetragen!Seit dem 27. Mai 2022 werden keine IVD gemäß IVDD (98/79/EG) mehr auf dem Schweizer Markt neu zugelassen. Noch auf dem Markt befindliche IVDs (Legacy Devices) dürfen keine signifikanten Änderungen erfahren!
Fazit
Nutzen Sie die verbleibende Zeit, sich mit den neuen Regulierungen auseinanderzusetzen, und stellen Sie sicher, dass Sie alle erforderlichen Schritte unternehmen, um rechtzeitig Konformität zu erreichen.Verlieren Sie die Übergangsfristen nicht aus den Augen. Die Fristen für die Benennung eines CH-Rep laufen bereits im März für Klasse C- & D-Produkte und im Juli für Klasse A-Produkte ab.Detailliertere Informationen finden Sie auch auf der Seite Swissmedic.ch im Merkblatt "FAQ - Meldung von In-vitro-Diagnostika".Sie benötigen Unterstützung bei der Erstellung und Einreichung der geforderten Dokumente bei der Swissmedic? Oder suchen Sie nach einem CH-Rep, um Ihre IVDs auf dem Schweizer Markt zuzulassen? Wir erleichtern Ihnen den Zugang zum Schweizer Markt – und beantworten auch gerne weitere Fragen zur Zulassung Ihrer IVDs. Wir freuen uns, von Ihnen zu hören!Beste Grüße
Unsere Blogbeiträge werden mit höchster Sorgfalt recherchiert und erstellt, sind jedoch lediglich Momentaufnahmen in der Regulatorik, und diese ist in stetem Wandel. Wir gewährleisten nicht, dass ältere Inhalte noch aktuell und aussagekräftig sind. Wenn Sie nicht sicher sind, ob der Beitrag, den Sie auf dieser Seite gelesen haben, noch dem aktuellen Stand der Regulierung entspricht, nehmen Sie bitte Kontakt zu uns auf: Wir ordnen Ihr Thema schnell in den aktuellen Kontext ein.
Regulatory Affairs Expert



