
Die Äquivalenz von Medizinprodukten unter der MDR: Ein Leitfaden
23.04.2024
Die Äquivalenz von Medizinprodukten gemäß den Vorgaben der Medical Device Regulation (EU) 2017/745 (MDR) ist ein wichtiger Aspekt, der Hersteller vor große Herausforderungen stellen kann. Besonders im Kontext der klinischen Bewertung unter der MDR wird deutlich, wie entscheidend die adäquate Nachweisführung von Gleichartigkeit ist. Die Benannten Stellen betonen die Bedeutung dieses Prozesses, der nicht nur zur Sicherstellung der Konformität beiträgt, sondern auch Zeit- und Kostenersparnisse ermöglicht.Doch wie genau wird die Gleichartigkeit nach MDR nachgewiesen? Und welche Hürden und Fallstricke sind dabei zu beachten? Im Folgenden wird ein umfassender Überblick über die Anforderungen, Prozesse und Implikationen der Gleichartigkeitsnachweisführung gemäß MDR gegeben.Hinweis: Die Benannten Stellen nennen die Verwirrung um gleichartige Produkte als einen Stolperstein auf dem Weg zur Konformität mit der MDR, an dem viele Hersteller scheitern. Unter der MDD wurde der Nachweis von Gleichartigkeit nach Anhang A1 der MEDDEV 2.7.1 rev. 4 durchgeführt, die MDR verschärft die Anforderungen im Anhang XIV und die Medical Device Coordination Group trägt in ihrer Guidance MDCG 2020-5 zur weiteren Aufklärung bei.
Wozu die Gleichartigkeit nachweisen
Für viele Medizinprodukte sind klinische Daten erforderlich, um die Konformität mit den relevanten grundlegenden Sicherheits- und Leistungsanforderungen gemäß Anhang I der MDR nachzuweisen. Wenn die klinische Bewertung nicht auf ausreichenden klinischen Daten beruht, sollen diese aus der Anwendung des Medizinprodukts im Rahmen seiner Zweckbestimmung generiert werden.Eine klinische Prüfung kann zeit- und kostenaufwändig sein und erfordert zusätzliche Kompetenzen, die im Unternehmen möglicherweise gar nicht vorhanden sind, z.B. im Design, in der Definition der Endpunkte, der Sicherung der Datenqualität und der Auswertung der Ergebnisse.Eine mögliche Lösung ist die Feststellung der Gleichartigkeit. Ist diese nachgewiesen, dürfen gemäß Anhang XIV, Abschnitt 3 der MDR die klinischen Daten des gleichartigen Produkts für die klinische Bewertung verwendet werden.Dieser Nachweis ist jedoch nicht immer einfach.Wichtig: durch Feststellung der Gleichartigkeit wird nur die Übertragung der klinischen Daten ermöglicht. Die nicht-klinischen Daten, wie Biokompatibilität, mechanische und funktionelle Tests, Benchmark-Tests und Tierversuche sind hier nicht inbegriffen.
Wie gelingt der Nachweis der Gleichartigkeit?
Bei der klinischen Bewertung von implantierbaren Produkten und Produkten der Klasse III ist die größte Hürde der im Artikel 61(5) geforderte vertraglich geregelte uneingeschränkte Zugang zur technischen Dokumentation des Medizinprodukts, mit dem die Gleichartigkeit nachgewiesen werden soll.Da Wettbewerber selten zu einem solchen Informationsaustausch bereit sind, bleibt diese Möglichkeit den Herstellern der meisten implantierbaren Medizinprodukte und/oder Medizinprodukte der Risikoklasse III verwehrt. Eine Ausnahme besteht, wenn, wie in Artikel 61(4) beschrieben, die Gleichartigkeit mit einem Produkt desselben Herstellers nachgewiesen werden soll.Für die Feststellung der Gleichartigkeit mit anderen als Klasse III oder implantierbaren Medizinprodukten ist zwar keinen Vertrag zwischen Herstellern erforderlich, der Zugang zu Informationen über das gleichartige Produkt sollte jedoch überprüft werden. Die Quellen sind der klinischen Bewertung beizufügen.Die Gleichartigkeit wird gemäß Anhang XIV Abschnitt 3 der MDR nachgewiesen, die Anforderungen des Anhang A1 der MEDDEV 2.7.1 rev. 4 sind unter MDR nicht anwendbar (s.o.). Die zu vergleichenden Merkmale sind in der folgenden Übersichtstabelle zusammengefasst.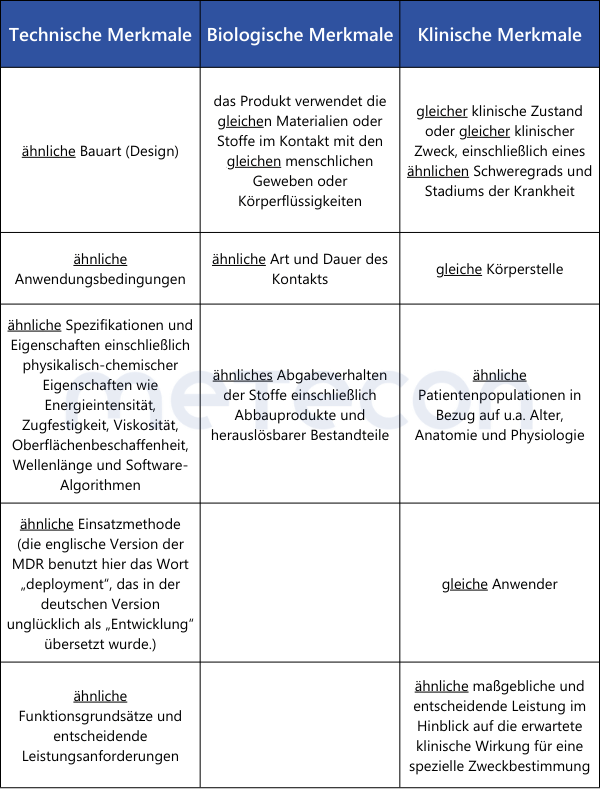
Was ist zu beachten?
Wie aus der Übersichtstabelle ersichtlich, müssen einige Punkte tatsächlich "gleich" sein, damit die Gleichartigkeit zweier Medizinprodukten korrekt nachgewiesen werden kann. Wird hier ein Unterschied festgestellt, ist die Gleichartigkeit nicht gegeben. Bei den übrigen Punkten, in denen nur eine Ähnlichkeit gefordert ist, müssen alle Unterschiede einzeln bewertet werden. Es ist wissenschaftlich zu begründen, ob diese Unterschiede signifikante Auswirkungen auf die klinische Leistung oder Sicherheit der Medizinprodukte haben.Für den Nachweis der Gleichartigkeit zweier Medizinprodukte ohne medizinischen Verwendungszweck (Beispiele aus Anhang XVI der MDR: Kontaktlinsen, Geräte zur Liposuktion, Lipolyse oder Lipoplastie) können die Erläuterungen aus MDCG 2023-6 herangezogen werden: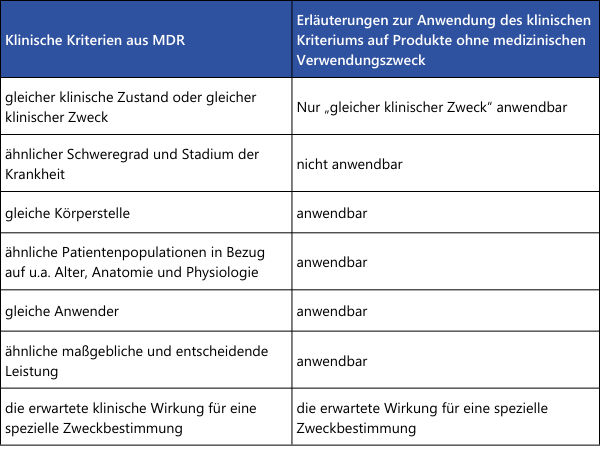
Was geht nicht?
Produkte mit unterschiedlicher Zweckbestimmung
Der Nachweis der Gleichwertigkeit kann nicht zwischen einem Produkt ohne medizinischen Verwendungszweck und einem vergleichbaren Produkt mit medizinischem Verwendungszweck erbracht werden.Wenn ein Produkt ohne medizinischen Verwendungszweck mit einem Produkt verglichen wird, das sowohl über eine medizinischen als auch einen nicht-medizinischen Verwendungszweck verfügt, so dürfen nur die Merkmale, die auf den nicht-medizinischen Verwendungszweck bezogen sind, betrachtet werden. Dementsprechend dürfen auch nach Feststellung der Gleichartigkeit nur diejenigen klinischen Daten übernommen werden, die mit der nichtmedizinischen Verwendung verbunden sind.Ähnliche Medizinprodukte
Ein in der Praxis häufig auftretender Fehler ist die Verwendung von klinischen Daten eines ähnlichen Medizinprodukts ohne Nachweis der Gleichartigkeit. Dies führt zu einer Abweichung bei der Konformitätsbewertung. Hier muss eine klare Abgrenzung getroffen werden. MDCG 2020-6 bezeichnet ähnliche Produkte als Produkte, die zu einer generischen Gruppe gehören und verweist auf die Definition aus Artikel 2(7) der MDR, wonach eine "generische Produktgruppe" eine Gruppe von Produkten mit gleicher oder ähnlicher Zweckbestimmungen oder mit technologischen Gemeinsamkeiten ist, die allgemein, also ohne Berücksichtigung spezifischer Merkmale, klassifiziert werden können.Auf der Grundlage klinischer Daten, die mit solchen Produkten generiert wurden, können Akzeptanzkriterien für die klinische Leistung oder Sicherheit generischer Medizinprodukte nach dem Stand der Technik dargestellt werden. Die Informationen aus der Fachliteratur über Designentwicklung, Sicherheits- und Leistungsprofil können bei der Beurteilung helfen, , ob es sich um eine bewährte (engl. "well-established") Technologie handelt.Bei Medizinprodukten, die auf einer bewährten Technologie beruhen, können klinische Daten ähnlicher Produkte unterstützend für den Nachweis der Konformität mit den relevanten grundlegenden Sicherheits- und Leistungsanforderungen herangezogen werden. Dabei bedeutet "unterstützend", dass der Nachweis eines Leistungs- oder Sicherheitsaspekts nicht ausschließlich auf den Daten ähnlicher Produkte beruhen darf.Predicate devices
Übrigens: Die MDR ist nicht das einzige Regelwerk für Medizinprodukte, in dem der Begriff "Gleichartigkeit" oder "Äquivalenz" definiert wird, und auch außerhalb der Europäischen Union wird dieses Konzept verwendet, wenn auch in unterschiedlicher Weise. Ein Beispiel hierfür ist das "predicate device", das auf "substantial equivalence" beruht und den Eckpfeiler der 510(k)-Verfahrens der US-amerikanischen FDA bildet. In solchen Fällen ist es wichtig zu verstehen, wie die Begriffe in den einzelnen Rechtsordnungen definiert und verwendet werden, um sicherzustellen, dass Sie jeweils konform handeln.Wie geht es weiter?
Gleichartige bzw. ähnliche Produkte, deren klinische Daten für die Konformitätsbewertung eines Produkts verwendet wurden, müssen im Rahmen von Maßnahmen der Post-Market Clinical Follow-up (PMCF) weiter beobachtet werden. Dies spiegelt sich in den von der MDCG vorgeschlagenen Vorlagen für den PMCF-Plan (MDCG 2020-7) und den PMCF-Bericht (MDCG 2020-8) wider. Damit soll eine kontinuierliche Aktualisierung des Nutzen-Risiko-Verhältnisses gewährleistet werden.Sie haben Fragen zur Äquivalenz von Medizinprodukten, zur klinischen Bewertung oder zu anderen Aspekten rund um die klinische Anwendung Ihres Medizinprodukts? Kontaktieren Sie mich gerne. Gemeinsam mit meinen Kolleginnen und Kollegen aus dem Bereich Clinical Affairs unterstütze ich Sie gerne in allen Belangen.
Wozu die Gleichartigkeit nachweisen
Für viele Medizinprodukte sind klinische Daten erforderlich, um die Konformität mit den relevanten grundlegenden Sicherheits- und Leistungsanforderungen gemäß Anhang I der MDR nachzuweisen. Wenn die klinische Bewertung nicht auf ausreichenden klinischen Daten beruht, sollen diese aus der Anwendung des Medizinprodukts im Rahmen seiner Zweckbestimmung generiert werden.Eine klinische Prüfung kann zeit- und kostenaufwändig sein und erfordert zusätzliche Kompetenzen, die im Unternehmen möglicherweise gar nicht vorhanden sind, z.B. im Design, in der Definition der Endpunkte, der Sicherung der Datenqualität und der Auswertung der Ergebnisse.Eine mögliche Lösung ist die Feststellung der Gleichartigkeit. Ist diese nachgewiesen, dürfen gemäß Anhang XIV, Abschnitt 3 der MDR die klinischen Daten des gleichartigen Produkts für die klinische Bewertung verwendet werden.Dieser Nachweis ist jedoch nicht immer einfach.Wichtig: durch Feststellung der Gleichartigkeit wird nur die Übertragung der klinischen Daten ermöglicht. Die nicht-klinischen Daten, wie Biokompatibilität, mechanische und funktionelle Tests, Benchmark-Tests und Tierversuche sind hier nicht inbegriffen.Wie gelingt der Nachweis der Gleichartigkeit?
Bei der klinischen Bewertung von implantierbaren Produkten und Produkten der Klasse III ist die größte Hürde der im Artikel 61(5) geforderte vertraglich geregelte uneingeschränkte Zugang zur technischen Dokumentation des Medizinprodukts, mit dem die Gleichartigkeit nachgewiesen werden soll.Da Wettbewerber selten zu einem solchen Informationsaustausch bereit sind, bleibt diese Möglichkeit den Herstellern der meisten implantierbaren Medizinprodukte und/oder Medizinprodukte der Risikoklasse III verwehrt. Eine Ausnahme besteht, wenn, wie in Artikel 61(4) beschrieben, die Gleichartigkeit mit einem Produkt desselben Herstellers nachgewiesen werden soll.Für die Feststellung der Gleichartigkeit mit anderen als Klasse III oder implantierbaren Medizinprodukten ist zwar keinen Vertrag zwischen Herstellern erforderlich, der Zugang zu Informationen über das gleichartige Produkt sollte jedoch überprüft werden. Die Quellen sind der klinischen Bewertung beizufügen.Die Gleichartigkeit wird gemäß Anhang XIV Abschnitt 3 der MDR nachgewiesen, die Anforderungen des Anhang A1 der MEDDEV 2.7.1 rev. 4 sind unter MDR nicht anwendbar (s.o.). Die zu vergleichenden Merkmale sind in der folgenden Übersichtstabelle zusammengefasst.Was ist zu beachten?
Wie aus der Übersichtstabelle ersichtlich, müssen einige Punkte tatsächlich "gleich" sein, damit die Gleichartigkeit zweier Medizinprodukten korrekt nachgewiesen werden kann. Wird hier ein Unterschied festgestellt, ist die Gleichartigkeit nicht gegeben. Bei den übrigen Punkten, in denen nur eine Ähnlichkeit gefordert ist, müssen alle Unterschiede einzeln bewertet werden. Es ist wissenschaftlich zu begründen, ob diese Unterschiede signifikante Auswirkungen auf die klinische Leistung oder Sicherheit der Medizinprodukte haben.Für den Nachweis der Gleichartigkeit zweier Medizinprodukte ohne medizinischen Verwendungszweck (Beispiele aus Anhang XVI der MDR: Kontaktlinsen, Geräte zur Liposuktion, Lipolyse oder Lipoplastie) können die Erläuterungen aus MDCG 2023-6 herangezogen werden:
 der MDCG")

