
Leitfaden Prozessvalidierung Teil 2 - Ruhige Fahrt statt stürmischer See
21.05.2024
Bereit für einen weiteren Ausflug in die Welt der Prozessvalidierung? Im Leitfaden Prozessvalidierung Teil 1 haben wir erörtert, welche Anforderungen Anhang I (Grundlegende Sicherheits- und Leistungsanforderungen (GSPR)) der Medical Device Regulation (EU) 2017/745 (MDR) und die DIN EN ISO 13485:2021-12 an die Validierung in der Produktion stellen, welche Alternativen es gibt, und wie die Entscheidungsfindung idealerweise abläuft. Im zweiten Teil wenden wir nun anhand eines Beispiel- Prozesses die vorgestellten Konzepte an und gehen detaillierter darauf ein, wie Sie eine Validierung konkret umsetzen können.Eine wichtige Anmerkung vorab: Für einige Bereiche existieren eigene normative Vorgaben. Sofern für Ihr Produkt durch Normen geregelt ist, wie Validierungen z.B. von Reinigung, Desinfektion, Sterilisation, Verpackung oder Aufbereitung durchgeführt werden sollen, so sind diese selbstverständlich zu beachten.
Schritt 1: Den Prozess beschreiben
Wir bedienen uns erneut des Beispiels aus unserem ersten Beitrag zum Thema: Es geht um die Mitarbeiterin Vera Alid der Firma KOMPONENTEN-KÖNIGE und um einen Abfüll- und Verpackungsprozess, für den vom Kunden, einem Hersteller im Bereich Medizintechnik, Validierungsunterlagen angefordert wurden. V. Alid nimmt sich die Arbeitsanweisungen für die Produkte und spricht mit dem Fertigungsleiter, um sich einen Überblick über die Prozesse zu machen, die für den Hersteller Medizinprodukte GmbH durchgeführt werden.Die folgende Beschreibung ist das Ergebnis ihrer Recherche:
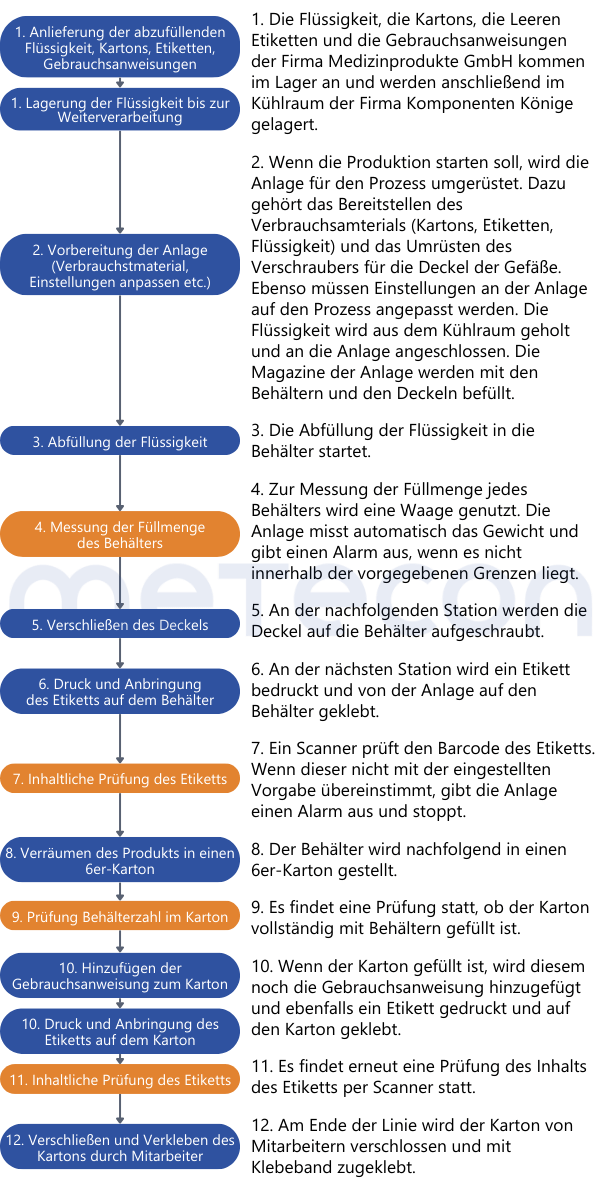
Schritt 2: Den Prozess analysieren
Nachdem feststeht, wie der Prozess durchgeführt wird, kann mit seiner Analyse begonnen werden. Da eine potenzielle Validierung im Raum steht, muss genau überlegt werden, welche Eigenschaften des Produktes relevant für Sicherheit und Leistung sind, wie sie geprüft werden und ob diese Prüfung als 100 %-Verifizierung eingestuft werden kann. Dazu hilft es, die Prozessergebnisse bzw. Produkteigenschaften aufzulisten. Je Eigenschaft kann mithilfe der Prozessbeschreibung eingeschätzt werden, ob und wie diese verifiziert werden. Eine solche Tabelle könnte beispielhaft wie folgt aussehen:

Die Flüssigkeit in den Behältern soll aus diesen nicht entweichen können und andererseits soll diese auch nicht von außen verunreinigt werden können. Daraus ergibt sich die Anforderung, dass die Behälter dicht sein müssen. Im Rahmen des Prozesses findet keine Überprüfung dieser Eigenschaft statt. Da die Dichtigkeit des Behälters als relevant für Sicherheit und Leistung des Produkts eingestuft wurde muss sie in der Validierung betrachtet werden.Aus der Messung des Gewichts lässt sich schließen, dass es eine Anforderung an die Füllmenge eines jeden Behälters gibt. Die Eigenschaft "Füllmenge" wird für jeden Behälter durch die Anlage geprüft. Für diese Eigenschaft liegt also eine 100 % Verifizierung vor. Damit muss diese Eigenschaft in einer potenziellen Validierung nicht betrachtet werden. Allerdings sollte, wie im vorherigen Beitrag angesprochen, diese Entscheidung begründet dokumentiert werden.Die Farbe der Behälter wurde als nicht relevant für die Sicherheit und Leistung des Produkts eingestuft. Ob und wie Sie überprüft wird ist in dem Kontext der Entscheidung für die Validierung also nicht weiter relevant.Die Informationen auf dem Etikett wurden als relevant für die Sicherheit und Leistung des Produkts bewertet. Ein in die Anlage eingebauter Scanner überprüft diese einzeln. Bei einer Abweichung von den Soll-Vorgaben stoppt die Anlage und gibt einen Alarm aus. Es ist also nicht nur sichergestellt, dass geprüft wird (100 % Verifizierung). Es ist auch eine Maßnahme implementiert, die angemessen das Risiko reduziert, dass Produkte mit fehlerhaften Etiketten weiterverarbeitet werden. Es muss allerdings ein Nachweis existieren, dass diese Maßnahme auch korrekt funktioniert (Stichwort Qualifizierung).Praxis Tipp: Als Lieferant für Hersteller von Medizinprodukten müssen Sie die Bewertung der Relevanz der Eigenschaft für Sicherheit und Leistung mit dem Hersteller zusammen durchführen. Als Hersteller ist es wichtig, dass Produktexperten bei der Entscheidungsfindung dabei sind, die über ein ausreichendes Produktwissen verfügen.
Die Risikoanalyse
Die Analyse des Prozesses kann mithilfe von Risikoanalysemethoden erfolgen. Diese bieten auch einen Nachweis für die Dokumentation, wie die Entscheidung zur Prüfung und zu deren Intensität getroffen wurde (Stichwort Statistik).Damit es nicht zu Verwirrungen kommt, wenn in diesem Beitrag von Risikoanalyse gesprochen wird, hier eine kleine Anmerkung: Für Prozessvalidierungen müssen keine Risikoanalysen nach ISO 14971 durchgeführt werden. Sie sind also frei in der Wahl der Risikoanalysemethode. Wichtig hierbei ist, dass auch die Entdeckungswahrscheinlichkeit von Fehlern in der Risikoanalyse dargestellt werden kann. Im Rahmen der Risikoanalyse sollte auch klar werden welche Prozessparameter vorhanden sind und welche davon kritisch sind. Der Begriff Parameter muss sich nicht allein auf Anlageneinstellungen beziehen, sondern kann für alle Prozessvariablen genutzt werden.Nach der Durchführung einer Risikoanalyse kann mit der Planung begonnen werden, wie nachgewiesen wird, dass die identifizierten Risiken mitigiert wurden. Damit kommen wir nun zum nächsten Schritt:
Funktionsqualifizierung – Operational Qualification (OQ)
Die Funktionsqualifizierung oder auch Operational Qualification (OQ) ist ein Teil einer Prozessvalidierung und wird gemäß GHTF definiert als Feststellung durch objektive Beweise, dass ein Prozess durchgängig ein Ergebnis oder Produkt erzeugt, das den vorher festgelegten Anforderungen entspricht ("establishing by objective evidence that a process consistently produces a result or product meeting its predetermined requirements"). Diese Definition schließt ein, dass nachgewiesen werden muss, dass der Prozess im "worst case", also unter den denkbar schlechtest möglichen, aber erlaubten Bedingungen noch die geforderten Prozessergebnisse produziert.Was für einen Prozess die jeweiligen Worst-Case-Bedingungen sind, muss jeweils individuell festgelegt werden. Typische Beispiele sind: kleinste oder größte (Verpackungs-) Variante, minimale odermaximale Prozessgeschwindigkeit, Variante mit unvorteilhaften Materialien, niedrigste oder höchste Temperatur.Es kann für einen Parameter auch mehrere Worst-Case-Bedingungen geben. Ein gutes Beispiel hierfür sind untere und obere Temperaturgrenzen beim Siegeln von Verpackungen: Beim unteren Grenzwert könnte die Siegelung undicht sein oder eine zu geringe Festigkeit aufweisen. Beim oberen Grenzwert könnte ggf. das Material beschädigt werden.BeispielAn der Anlage kann nicht die Füllmenge der Behälter sondern lediglich der Parameter "Abfülldauer" eingestellt werden. (Annahme: Es findet auch keine 100 % Prüfung der Füllmenge an der Anlage statt) Die Füllmenge des Behälters hat eine Spezifikation mit einer unteren und oberen Grenze. Die Abfülldauer kann zu kurz oder zu lang eingestellt werden, sodass die Spezifikation der Füllmenge nicht eingehalten wird. Wenn nun an der Anlage ein Bereich von 2-4 Sekunden als Einstellung erlaubt ist, so müssten sowohl die untere und als auch die obere Grenze dieser Einstellung bei einer Validierung betrachtet werden.Es müssen grundsätzlich alle Worst-Case-Fälle in der Validierung betrachtet werden. Bei Prozessen, die mit mehreren unterschiedlichen Input-Variablen (Material, Größen, etc.) durchgeführt werden, ist die Darstellung der Worst-Case-Kombinationen in einer Matrix hilfreich. So ist nachvollziehbar dokumentiert, warum die überprüften Worst-Case-Kombinationen ausgewählt wurden. Aber nicht nur bei der erstmaligen Bestimmung ist diese Matrix hilfreich. Auch wenn Änderungen am Prozess bzw. an den Variablen vorgenommen werden, kann diese Matrix genutzt werden, um die Notwendigkeit erneuter OQ-Tests zu bestimmen.Parameter, für die während der Produktion nur eine Vorgabe vorhanden ist, z.B. Anlagengeschwindigkeit, werden während der OQ konstant auf diesem Wert belassen. Die Einstellungen bei der OQ werden nicht außerhalb von dem variiert, was auch für den Prozess vorgeschrieben ist. In so einem Fall gibt es keinen speziellen „worst case“.
Einschub: Prozessentwicklung
Die OQ soll nachweisen, dass der Prozess an den festgelegten Grenzen noch akzeptable Ergebnisse produziert. Doch woher sind die Grenzen bekannt?Eigentlich sollten sie im Rahmen der Prozessentwicklung bestimmt worden sein. Doch wie gehen Sie vor, wenn solche Entwicklungsberichte nicht vorliegen, der Prozess aber schon über einen längeren Zeitraum durchgeführt wird?Mitarbeiterin V. Alid kann sich das Gesicht ihres Chefs gut ausmalen, wenn sie ihm vorschlägt, Entwicklungstests durchzuführen. Eine Möglichkeit in so einer Situation ist es, die aktuellen Vorgaben der Grenzen zu nutzen. Hierbei sollte in Erfahrung gebracht werden, ob in der Vergangenheit die Worst-Case-Kombinationen z. B. schnellste Geschwindigkeit und niedrigste Temperatur bei einem Siegelprozess verwendet wurden, und ob es dabei Probleme gab. In diesem Fall könnte es auch in der OQ Probleme geben, die Zeit und Ressourcen kosten.Grundsätzlich sollten Sie prüfen, ob die vorhandenen Arbeitsanweisungen präzise genug die Vorgaben für wichtige Parameter nennen und ob sie im Einklang mit den Prozessparametergrenzen stehen, die in der OQ genannt werden. Prozesskenntnis ist für die Planung der OQ unglaublich wichtig und kann Ihnen eine wiederholte Durchführung von Tests ersparen.
Leistungsqualifizierung – Performance Qualification (PQ)
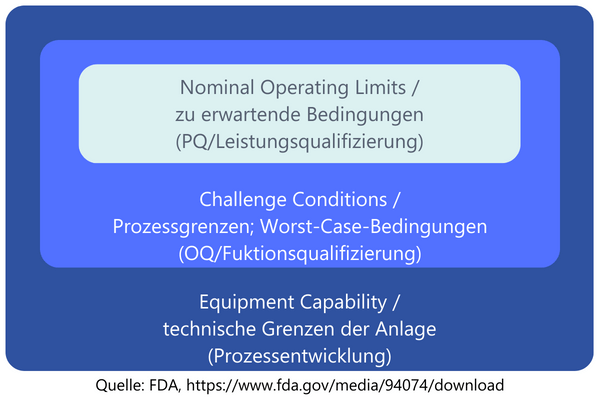
Bei der Leistungsqualifizierung bzw. Performance Qualification soll der Nachweis erbracht werden, dass das Verfahren unter den zu erwartenden Bedingungen durchgängig ein Produkt erzeugt, das alle vorher festgelegten Anforderungen erfüllt (GHTF-Definition). Die zu erwartenden Bedingungen sind hierbei das, was für den Prozess ermittelt werden muss.Beispiele für Variationen der Bedingungen sind einerseits bei den direkten Input-Variablen zu finden, andererseits aber auch bei den Bedingungen während der Durchführung. Beispiele können sein: unterschiedliche Materialchargen, Variationen der Umgebungsbedingungen (Temperatur, Luftfeuchte), Variation des durchführenden Personals, Umrüstvorgänge an den Anlagen, die den Prozess durchführen, generelles Anfahren und Stoppen der Anlage/des Prozesses und Verschleiß von genutzten Werkzeugen.Welche möglichen Variationen es gibt und wie diese im Kontext des durchzuführenden Prozesses zu bewerten sind, sollte in einer Risikoanalyse bewertet werden. So können Sie auch aufzeigen, dass Sie die Variationen identifiziert haben, eine weitere Untersuchung der Effekte im Rahmen einer PQ aber nicht notwendig ist. Dafür müssen Sie eine ausreichende Argumentationsgrundlage besitzen.Bei der OQ war die Anzahl der durchzuführenden Prozessläufe durch die Anzahl der Worst-Case-Szenarien vorgegeben. Bei der PQ ist dies auf den ersten Blick etwas schwieriger zu entscheiden. Oft werden einfach drei Chargen hergestellt. Dies war im Bereich der Arzneimittelherstellung eine lange Zeit Usus. In der aktuellen Regulatorik für Medizinprodukte (MDSAP AA, ISO 13485, GHTF) lässt sich eine solche Vorgabe oder Empfehlung allerdings nicht finden.Auch hier können aber die Zauberworte "risikobasierter Ansatz" weiterhelfen, denn das eigentliche Ziel ist es, mit den Chargen die Variation der Bedingungen "einzufangen" und dann die Ergebnisse dieser Variationen zu untersuchen. Ziel ist es am Ende nachzuweisen, dass das Prozessergebnis die geforderten Spezifikationen erfüllt. Wie viele Chargen für diese Bedingungen benötigt werden, können Sie im Rahmen der Risikoanalyse oder der PQ-Planung festlegen. Wenn Sie sich unsicher sind, ist die Durchführung von drei Chargen sicher eine gute Richtschnur zur Orientierung.Praxis Tipp: Angenommen ein zu validierender Prozess läuft schon seit einigen Jahren ohne Änderungen. In solchen Fällen könnten Sie prüfen, ob eine ausreichende Datenmenge vorliegt, um die PQ retrospektiv durchzuführen. Daten, die Sie hierfür benötigen, wären z. B. Nachweise über Prozessparameter oder Chargen der Eingangsmaterialien, die während der Herstellung genutzt wurden. In einer retrospektiven Validierung könnten Sie eingangs mögliche Variationen der zu erwartenenden Bedingungen nennen und anhand der vorliegenden Daten aufzeigen, dass diese Variationen in der Vergangenheit aufgetreten sind und dabei Produkte hergestellt wurden, die den Spezifikationen entsprechen. Wichtig ist dabei, auch aufgetretene Nicht-Konformitäten des Prozesses zu bewerten und so zu zeigen, dass auch unvorteilhafte Daten geprüft und bewertet wurden.Die folgende Grafik (angelehnt an eine Darstellung der US-amerikanischen Food and Drug Administration (FDA)) zeigt die Betrachtungsweisen von Prozessentwicklung, Operational Qualification und Performance Qualification. Während bei der Entwicklung noch sehr grob geprüft wird, was möglich ist, und das auch teilweise an den technischen Grenzen der Anlage, wird sich mit OQ und PQ immer mehr an die „normalen“ Bedingungen während der Produktion angenähert.
Allgemeine Dokumentation der Tätigkeiten
Sowohl für die Funktionsqualifizierung (OQ) als auch für die Leistungsqualifizierung (PQ) müssen Pläne und Berichte geschrieben werden. Aus den Berichten sollte klar hervorgehen, dass die im Plan erwähnten Aktivitäten durchgeführt wurden. Abweichungen von der Planung müssen begründet werden.Je Validierungsprojekt können übergeordnete Dokumente erstellt werden, die auch als Kontrollmechanismus hilfreich sind, um die Erbringung notwendiger Nachweise übersichtlich zu dokumentieren. Ebenfalls kann hier der Prozess inkl. der Ergebnisse beschrieben werden. Eine solche Darstellung ermöglicht im Audit einen einfachen Einstieg in das Validierungsprojekt für Sie, aber auch für den Auditor. So wird der gesamte Kontext klarer und die nachfolgenden Dokumente der OQ und PQ können kürzer ausfallen. Zudem kann in einem abschließenden „Validierungsprojektabschlussbericht“ aufgezeigt werden welche Dokumente zum Projekt gehören, und es kann kontrolliert und herausgestellt werden, dass alle geplanten Tätigkeiten abgeschlossen sind.Insbesondere bei größeren Projekten helfen solche übergeordneten Dokumente, einen Überblick zu behalten und diesen im Nachhinein auch Auditoren zu vermitteln.Praxis Tipp: Wenn bei Ihnen noch keine Validierung vorliegt, diese aber notwendig ist, dann ist dies ein geeigneter Zeitpunkt, um mögliche bereits angedachte Änderungen an Ihrem Prozess durchzuführen. Änderungen, nachdem die Validierung durchgeführt wurde, können eine Revalidierung zur Folge haben.
Bei Änderungen am Prozess können Revalidierungen notwendig werden. Es kann Änderungen am Material, an der verwendeten Ausrüstung, am Ort der Durchführung des Prozesses oder den Prozessparametern geben. Bei Planung einer Änderung muss daher bewertet werden, ob diese Änderung einen Einfluss auf den Validierungsstatus des Prozesses hat. In jedem Fall ist es wichtig die Entscheidung begründet zu dokumentieren und Fachexperten in die Bewertung einzubeziehen. Teilweise kann es ausreichen, nur Teile der Validierung nachzuholen. Vielleicht sind nur Teile des Prozesses betroffen. Wenn eine gute Risikoanalyse des Prozesses vorliegt, hilft Ihnen dies ungemein bei der Bewertung solche Änderungen.Was heißt das nun für unseren Abfüllprozess? Angenommen, es findet keine 100 % Verifizierung der Füllmenge statt. Die Spezifikation für die Füllmenge des Behälters soll angepasst und die obere Grenze erhöht werden. Da die Füllmenge durch den Parameter der Abfülldauer beeinflusst wird, wird also dessen obere Grenze erhöht. Tests in der OQ, bei denen mit der unteren Grenze des Parameters gearbeitet wurde, müssen nicht wiederholt werden, da dieser nicht angepasst wird. Tests für die Dichtigkeit könnten ggf. auch weggelassen werden, wenn begründet wird, dass eine höhere Füllmenge keinen Einfluss auf diesen Parameter hat. Somit kann es sein, dass nur noch Tests mit der oberen Parametergrenze für die Abfülldauer durchgeführt werden müssen.Auch für Prozessanpassungen ist es wichtig, eine Basis für die OQ zu schaffen. Welche und in welchem Umfang Prozessparameter bei Änderungen von Spezifikation oder Material geändert werden, ist meist nicht eindeutig. In solchen Situationen sind Versuche notwendig, die mit ausreichender Sicherheit Prozessparameter und Grenzen definieren, die für die OQ genutzt werden können.
Fazit:
Auch in diesem Beitrag war es unser Ziel, die Bedeutung von Prozess- und Produktkenntnissen für die Planung der Validierung herauszustellen Ihnen ist hoffentlich klarer geworden welche Aktivitäten hinter den Begriffen Operational Qualification (OQ) und Performane Qualification (PQ) stecken und wie Sie Ihre Kompetenz auf dem Gebiet auch Auditoren vermitteln können. Die Qualifizierung von Geräten, Equipment und Anlagen, einen weiteren wichtigen Teilaspekt der Prozessvalidierung, haben wir bisher nicht beleuchtet. Dies werden wir in einem künftigen Beitrag nachholen.Uns ist bewusst, dass der Themenkomplex Verifikation und Validierung bei der ersten Konfrontation viele Fragen aufwerfen kann. Das bestätigen uns neue Kunden immer wieder im Gespräch. Wir hoffen aber, dass wir Ihnen mit unserem Leitfaden Prozessvalidierung eine erste Hilfestellung bieten können.Wo Sie dann immer noch nicht weiterkommen, geben wir Ihnen natürlich gerne auch eine individuelle Rückmeldung auf Ihre Fragen.
 Leon Weißenhorn
Leon Weißenhorn
Schritt 1: Den Prozess beschreiben
Wir bedienen uns erneut des Beispiels aus unserem ersten Beitrag zum Thema: Es geht um die Mitarbeiterin Vera Alid der Firma KOMPONENTEN-KÖNIGE und um einen Abfüll- und Verpackungsprozess, für den vom Kunden, einem Hersteller im Bereich Medizintechnik, Validierungsunterlagen angefordert wurden. V. Alid nimmt sich die Arbeitsanweisungen für die Produkte und spricht mit dem Fertigungsleiter, um sich einen Überblick über die Prozesse zu machen, die für den Hersteller Medizinprodukte GmbH durchgeführt werden.Die folgende Beschreibung ist das Ergebnis ihrer Recherche:Schritt 2: Den Prozess analysieren
Nachdem feststeht, wie der Prozess durchgeführt wird, kann mit seiner Analyse begonnen werden. Da eine potenzielle Validierung im Raum steht, muss genau überlegt werden, welche Eigenschaften des Produktes relevant für Sicherheit und Leistung sind, wie sie geprüft werden und ob diese Prüfung als 100 %-Verifizierung eingestuft werden kann. Dazu hilft es, die Prozessergebnisse bzw. Produkteigenschaften aufzulisten. Je Eigenschaft kann mithilfe der Prozessbeschreibung eingeschätzt werden, ob und wie diese verifiziert werden. Eine solche Tabelle könnte beispielhaft wie folgt aussehen:Die Flüssigkeit in den Behältern soll aus diesen nicht entweichen können und andererseits soll diese auch nicht von außen verunreinigt werden können. Daraus ergibt sich die Anforderung, dass die Behälter dicht sein müssen. Im Rahmen des Prozesses findet keine Überprüfung dieser Eigenschaft statt. Da die Dichtigkeit des Behälters als relevant für Sicherheit und Leistung des Produkts eingestuft wurde muss sie in der Validierung betrachtet werden.Aus der Messung des Gewichts lässt sich schließen, dass es eine Anforderung an die Füllmenge eines jeden Behälters gibt. Die Eigenschaft "Füllmenge" wird für jeden Behälter durch die Anlage geprüft. Für diese Eigenschaft liegt also eine 100 % Verifizierung vor. Damit muss diese Eigenschaft in einer potenziellen Validierung nicht betrachtet werden. Allerdings sollte, wie im vorherigen Beitrag angesprochen, diese Entscheidung begründet dokumentiert werden.Die Farbe der Behälter wurde als nicht relevant für die Sicherheit und Leistung des Produkts eingestuft. Ob und wie Sie überprüft wird ist in dem Kontext der Entscheidung für die Validierung also nicht weiter relevant.Die Informationen auf dem Etikett wurden als relevant für die Sicherheit und Leistung des Produkts bewertet. Ein in die Anlage eingebauter Scanner überprüft diese einzeln. Bei einer Abweichung von den Soll-Vorgaben stoppt die Anlage und gibt einen Alarm aus. Es ist also nicht nur sichergestellt, dass geprüft wird (100 % Verifizierung). Es ist auch eine Maßnahme implementiert, die angemessen das Risiko reduziert, dass Produkte mit fehlerhaften Etiketten weiterverarbeitet werden. Es muss allerdings ein Nachweis existieren, dass diese Maßnahme auch korrekt funktioniert (Stichwort Qualifizierung).Praxis Tipp: Als Lieferant für Hersteller von Medizinprodukten müssen Sie die Bewertung der Relevanz der Eigenschaft für Sicherheit und Leistung mit dem Hersteller zusammen durchführen. Als Hersteller ist es wichtig, dass Produktexperten bei der Entscheidungsfindung dabei sind, die über ein ausreichendes Produktwissen verfügen.
Die Risikoanalyse
Die Analyse des Prozesses kann mithilfe von Risikoanalysemethoden erfolgen. Diese bieten auch einen Nachweis für die Dokumentation, wie die Entscheidung zur Prüfung und zu deren Intensität getroffen wurde (Stichwort Statistik).Damit es nicht zu Verwirrungen kommt, wenn in diesem Beitrag von Risikoanalyse gesprochen wird, hier eine kleine Anmerkung: Für Prozessvalidierungen müssen keine Risikoanalysen nach ISO 14971 durchgeführt werden. Sie sind also frei in der Wahl der Risikoanalysemethode. Wichtig hierbei ist, dass auch die Entdeckungswahrscheinlichkeit von Fehlern in der Risikoanalyse dargestellt werden kann. Im Rahmen der Risikoanalyse sollte auch klar werden welche Prozessparameter vorhanden sind und welche davon kritisch sind. Der Begriff Parameter muss sich nicht allein auf Anlageneinstellungen beziehen, sondern kann für alle Prozessvariablen genutzt werden.Nach der Durchführung einer Risikoanalyse kann mit der Planung begonnen werden, wie nachgewiesen wird, dass die identifizierten Risiken mitigiert wurden. Damit kommen wir nun zum nächsten Schritt:Funktionsqualifizierung – Operational Qualification (OQ)
Die Funktionsqualifizierung oder auch Operational Qualification (OQ) ist ein Teil einer Prozessvalidierung und wird gemäß GHTF definiert als Feststellung durch objektive Beweise, dass ein Prozess durchgängig ein Ergebnis oder Produkt erzeugt, das den vorher festgelegten Anforderungen entspricht ("establishing by objective evidence that a process consistently produces a result or product meeting its predetermined requirements"). Diese Definition schließt ein, dass nachgewiesen werden muss, dass der Prozess im "worst case", also unter den denkbar schlechtest möglichen, aber erlaubten Bedingungen noch die geforderten Prozessergebnisse produziert.Was für einen Prozess die jeweiligen Worst-Case-Bedingungen sind, muss jeweils individuell festgelegt werden. Typische Beispiele sind: kleinste oder größte (Verpackungs-) Variante, minimale odermaximale Prozessgeschwindigkeit, Variante mit unvorteilhaften Materialien, niedrigste oder höchste Temperatur.Es kann für einen Parameter auch mehrere Worst-Case-Bedingungen geben. Ein gutes Beispiel hierfür sind untere und obere Temperaturgrenzen beim Siegeln von Verpackungen: Beim unteren Grenzwert könnte die Siegelung undicht sein oder eine zu geringe Festigkeit aufweisen. Beim oberen Grenzwert könnte ggf. das Material beschädigt werden.BeispielAn der Anlage kann nicht die Füllmenge der Behälter sondern lediglich der Parameter "Abfülldauer" eingestellt werden. (Annahme: Es findet auch keine 100 % Prüfung der Füllmenge an der Anlage statt) Die Füllmenge des Behälters hat eine Spezifikation mit einer unteren und oberen Grenze. Die Abfülldauer kann zu kurz oder zu lang eingestellt werden, sodass die Spezifikation der Füllmenge nicht eingehalten wird. Wenn nun an der Anlage ein Bereich von 2-4 Sekunden als Einstellung erlaubt ist, so müssten sowohl die untere und als auch die obere Grenze dieser Einstellung bei einer Validierung betrachtet werden.Es müssen grundsätzlich alle Worst-Case-Fälle in der Validierung betrachtet werden. Bei Prozessen, die mit mehreren unterschiedlichen Input-Variablen (Material, Größen, etc.) durchgeführt werden, ist die Darstellung der Worst-Case-Kombinationen in einer Matrix hilfreich. So ist nachvollziehbar dokumentiert, warum die überprüften Worst-Case-Kombinationen ausgewählt wurden. Aber nicht nur bei der erstmaligen Bestimmung ist diese Matrix hilfreich. Auch wenn Änderungen am Prozess bzw. an den Variablen vorgenommen werden, kann diese Matrix genutzt werden, um die Notwendigkeit erneuter OQ-Tests zu bestimmen.Parameter, für die während der Produktion nur eine Vorgabe vorhanden ist, z.B. Anlagengeschwindigkeit, werden während der OQ konstant auf diesem Wert belassen. Die Einstellungen bei der OQ werden nicht außerhalb von dem variiert, was auch für den Prozess vorgeschrieben ist. In so einem Fall gibt es keinen speziellen „worst case“.Einschub: Prozessentwicklung
Die OQ soll nachweisen, dass der Prozess an den festgelegten Grenzen noch akzeptable Ergebnisse produziert. Doch woher sind die Grenzen bekannt?Eigentlich sollten sie im Rahmen der Prozessentwicklung bestimmt worden sein. Doch wie gehen Sie vor, wenn solche Entwicklungsberichte nicht vorliegen, der Prozess aber schon über einen längeren Zeitraum durchgeführt wird?Mitarbeiterin V. Alid kann sich das Gesicht ihres Chefs gut ausmalen, wenn sie ihm vorschlägt, Entwicklungstests durchzuführen. Eine Möglichkeit in so einer Situation ist es, die aktuellen Vorgaben der Grenzen zu nutzen. Hierbei sollte in Erfahrung gebracht werden, ob in der Vergangenheit die Worst-Case-Kombinationen z. B. schnellste Geschwindigkeit und niedrigste Temperatur bei einem Siegelprozess verwendet wurden, und ob es dabei Probleme gab. In diesem Fall könnte es auch in der OQ Probleme geben, die Zeit und Ressourcen kosten.Grundsätzlich sollten Sie prüfen, ob die vorhandenen Arbeitsanweisungen präzise genug die Vorgaben für wichtige Parameter nennen und ob sie im Einklang mit den Prozessparametergrenzen stehen, die in der OQ genannt werden. Prozesskenntnis ist für die Planung der OQ unglaublich wichtig und kann Ihnen eine wiederholte Durchführung von Tests ersparen.Leistungsqualifizierung – Performance Qualification (PQ)
Bei der Leistungsqualifizierung bzw. Performance Qualification soll der Nachweis erbracht werden, dass das Verfahren unter den zu erwartenden Bedingungen durchgängig ein Produkt erzeugt, das alle vorher festgelegten Anforderungen erfüllt (GHTF-Definition). Die zu erwartenden Bedingungen sind hierbei das, was für den Prozess ermittelt werden muss.Beispiele für Variationen der Bedingungen sind einerseits bei den direkten Input-Variablen zu finden, andererseits aber auch bei den Bedingungen während der Durchführung. Beispiele können sein: unterschiedliche Materialchargen, Variationen der Umgebungsbedingungen (Temperatur, Luftfeuchte), Variation des durchführenden Personals, Umrüstvorgänge an den Anlagen, die den Prozess durchführen, generelles Anfahren und Stoppen der Anlage/des Prozesses und Verschleiß von genutzten Werkzeugen.Welche möglichen Variationen es gibt und wie diese im Kontext des durchzuführenden Prozesses zu bewerten sind, sollte in einer Risikoanalyse bewertet werden. So können Sie auch aufzeigen, dass Sie die Variationen identifiziert haben, eine weitere Untersuchung der Effekte im Rahmen einer PQ aber nicht notwendig ist. Dafür müssen Sie eine ausreichende Argumentationsgrundlage besitzen.Bei der OQ war die Anzahl der durchzuführenden Prozessläufe durch die Anzahl der Worst-Case-Szenarien vorgegeben. Bei der PQ ist dies auf den ersten Blick etwas schwieriger zu entscheiden. Oft werden einfach drei Chargen hergestellt. Dies war im Bereich der Arzneimittelherstellung eine lange Zeit Usus. In der aktuellen Regulatorik für Medizinprodukte (MDSAP AA, ISO 13485, GHTF) lässt sich eine solche Vorgabe oder Empfehlung allerdings nicht finden.Auch hier können aber die Zauberworte "risikobasierter Ansatz" weiterhelfen, denn das eigentliche Ziel ist es, mit den Chargen die Variation der Bedingungen "einzufangen" und dann die Ergebnisse dieser Variationen zu untersuchen. Ziel ist es am Ende nachzuweisen, dass das Prozessergebnis die geforderten Spezifikationen erfüllt. Wie viele Chargen für diese Bedingungen benötigt werden, können Sie im Rahmen der Risikoanalyse oder der PQ-Planung festlegen. Wenn Sie sich unsicher sind, ist die Durchführung von drei Chargen sicher eine gute Richtschnur zur Orientierung.Praxis Tipp: Angenommen ein zu validierender Prozess läuft schon seit einigen Jahren ohne Änderungen. In solchen Fällen könnten Sie prüfen, ob eine ausreichende Datenmenge vorliegt, um die PQ retrospektiv durchzuführen. Daten, die Sie hierfür benötigen, wären z. B. Nachweise über Prozessparameter oder Chargen der Eingangsmaterialien, die während der Herstellung genutzt wurden. In einer retrospektiven Validierung könnten Sie eingangs mögliche Variationen der zu erwartenenden Bedingungen nennen und anhand der vorliegenden Daten aufzeigen, dass diese Variationen in der Vergangenheit aufgetreten sind und dabei Produkte hergestellt wurden, die den Spezifikationen entsprechen. Wichtig ist dabei, auch aufgetretene Nicht-Konformitäten des Prozesses zu bewerten und so zu zeigen, dass auch unvorteilhafte Daten geprüft und bewertet wurden.Die folgende Grafik (angelehnt an eine Darstellung der US-amerikanischen Food and Drug Administration (FDA)) zeigt die Betrachtungsweisen von Prozessentwicklung, Operational Qualification und Performance Qualification. Während bei der Entwicklung noch sehr grob geprüft wird, was möglich ist, und das auch teilweise an den technischen Grenzen der Anlage, wird sich mit OQ und PQ immer mehr an die „normalen“ Bedingungen während der Produktion angenähert.Allgemeine Dokumentation der Tätigkeiten
Sowohl für die Funktionsqualifizierung (OQ) als auch für die Leistungsqualifizierung (PQ) müssen Pläne und Berichte geschrieben werden. Aus den Berichten sollte klar hervorgehen, dass die im Plan erwähnten Aktivitäten durchgeführt wurden. Abweichungen von der Planung müssen begründet werden.Je Validierungsprojekt können übergeordnete Dokumente erstellt werden, die auch als Kontrollmechanismus hilfreich sind, um die Erbringung notwendiger Nachweise übersichtlich zu dokumentieren. Ebenfalls kann hier der Prozess inkl. der Ergebnisse beschrieben werden. Eine solche Darstellung ermöglicht im Audit einen einfachen Einstieg in das Validierungsprojekt für Sie, aber auch für den Auditor. So wird der gesamte Kontext klarer und die nachfolgenden Dokumente der OQ und PQ können kürzer ausfallen. Zudem kann in einem abschließenden „Validierungsprojektabschlussbericht“ aufgezeigt werden welche Dokumente zum Projekt gehören, und es kann kontrolliert und herausgestellt werden, dass alle geplanten Tätigkeiten abgeschlossen sind.Insbesondere bei größeren Projekten helfen solche übergeordneten Dokumente, einen Überblick zu behalten und diesen im Nachhinein auch Auditoren zu vermitteln.Praxis Tipp: Wenn bei Ihnen noch keine Validierung vorliegt, diese aber notwendig ist, dann ist dies ein geeigneter Zeitpunkt, um mögliche bereits angedachte Änderungen an Ihrem Prozess durchzuführen. Änderungen, nachdem die Validierung durchgeführt wurde, können eine Revalidierung zur Folge haben.Bei Änderungen am Prozess können Revalidierungen notwendig werden. Es kann Änderungen am Material, an der verwendeten Ausrüstung, am Ort der Durchführung des Prozesses oder den Prozessparametern geben. Bei Planung einer Änderung muss daher bewertet werden, ob diese Änderung einen Einfluss auf den Validierungsstatus des Prozesses hat. In jedem Fall ist es wichtig die Entscheidung begründet zu dokumentieren und Fachexperten in die Bewertung einzubeziehen. Teilweise kann es ausreichen, nur Teile der Validierung nachzuholen. Vielleicht sind nur Teile des Prozesses betroffen. Wenn eine gute Risikoanalyse des Prozesses vorliegt, hilft Ihnen dies ungemein bei der Bewertung solche Änderungen.Was heißt das nun für unseren Abfüllprozess? Angenommen, es findet keine 100 % Verifizierung der Füllmenge statt. Die Spezifikation für die Füllmenge des Behälters soll angepasst und die obere Grenze erhöht werden. Da die Füllmenge durch den Parameter der Abfülldauer beeinflusst wird, wird also dessen obere Grenze erhöht. Tests in der OQ, bei denen mit der unteren Grenze des Parameters gearbeitet wurde, müssen nicht wiederholt werden, da dieser nicht angepasst wird. Tests für die Dichtigkeit könnten ggf. auch weggelassen werden, wenn begründet wird, dass eine höhere Füllmenge keinen Einfluss auf diesen Parameter hat. Somit kann es sein, dass nur noch Tests mit der oberen Parametergrenze für die Abfülldauer durchgeführt werden müssen.Auch für Prozessanpassungen ist es wichtig, eine Basis für die OQ zu schaffen. Welche und in welchem Umfang Prozessparameter bei Änderungen von Spezifikation oder Material geändert werden, ist meist nicht eindeutig. In solchen Situationen sind Versuche notwendig, die mit ausreichender Sicherheit Prozessparameter und Grenzen definieren, die für die OQ genutzt werden können.
Fazit:
Auch in diesem Beitrag war es unser Ziel, die Bedeutung von Prozess- und Produktkenntnissen für die Planung der Validierung herauszustellen Ihnen ist hoffentlich klarer geworden welche Aktivitäten hinter den Begriffen Operational Qualification (OQ) und Performane Qualification (PQ) stecken und wie Sie Ihre Kompetenz auf dem Gebiet auch Auditoren vermitteln können. Die Qualifizierung von Geräten, Equipment und Anlagen, einen weiteren wichtigen Teilaspekt der Prozessvalidierung, haben wir bisher nicht beleuchtet. Dies werden wir in einem künftigen Beitrag nachholen.Uns ist bewusst, dass der Themenkomplex Verifikation und Validierung bei der ersten Konfrontation viele Fragen aufwerfen kann. Das bestätigen uns neue Kunden immer wieder im Gespräch. Wir hoffen aber, dass wir Ihnen mit unserem Leitfaden Prozessvalidierung eine erste Hilfestellung bieten können.Wo Sie dann immer noch nicht weiterkommen, geben wir Ihnen natürlich gerne auch eine individuelle Rückmeldung auf Ihre Fragen.
Regulatory Affairs & Technical Documentation


 der MDCG")