
Leitfaden Prozessvalidierung - Ruhige Fahrt statt stürmischer See
07.05.2024
Ein Deep Dive in die Prozessvalidierung: Wo ist eine Validierung in der Produktion notwendig, und wo sind Alternativen wie eine hundertprozentige Verifizierung des Ergebnisses denkbar? In diesem Beitrag geben wir Ihnen eine praktikable Hilfestellung für die Entscheidungsfindung an die Hand. Die beste Nachricht vorweg: Das Thema muss keine Leidensgeschichte sein – weder für Hersteller von Medizinprodukten noch für ihre Zulieferer.
PrologDas Unternehmen KOMPONENTEN-KÖNIGE ist seit vielen Jahren erfolgreich am Markt. Es hat die Räumlichkeiten, die Anlagen und das Know-how, um am Standort Flüssigkeiten in Behälter abzufüllen. Ein Kunde ist die Medizinprodukte GmbH, für die KOMPONENTEN-KÖNIGE seit fünf Jahren einen nahezu vollautomatischen Abfüll- und Verpackungsprozess durchführt.Letzte Woche hat die Medizinprodukte GmbH telefonisch angefragt, ob Prozessvalidierungsunterlagen vorhanden seien; sie würden zeitnah benötigt! Vera Alid, Mitarbeiterin bei KOMPONENTEN-KÖNIGE, hat versprochen, sich darum zu kümmern. Nach mehreren Gesprächen mit Kolleginnen und Kollegen schwirrt Vera der Kopf: Weiß sie überhaupt, wonach genau sie sucht? Allmählich zweifelt sie daran, denn die Aussagen der Befragten sind verwirrend und manchmal widersprüchlich:
Validierung in der Produktion -Teil 1: Die Suche nach dem Anfang
Zunächst muss eine ganz grundlegende Frage geklärt werden:
Was genau meint der Hersteller, wenn er eine Prozessvalidierung sehen will?
Gemäß der GHTF Guidance Quality Management Systems - Process Validation Guidance wird Prozessvalidierung wie folgt definiert: "establishing by objective evidence that a process consistently produces a result or product meeting its predetermined requirements." Es soll also durch objektive Nachweise festgestellt werden, dass ein Prozess kontinuierlich ein Ergebnis oder Produkt erzeugt, das seinen vorbestimmten Anforderungen entspricht. Diese Definition wurde auch von der US-amerikanischen Food and Drug Administration (FDA) in den 21 CFR Part 820.3 übernommen. Die Firma in unserem Fallbeispiel will also einen Nachweis für die zuvor genannte Aussage haben.In Anhang I (Grundlegenden Sicherheits- und Leistungsanforderungen (GSPR)) der Medical Device Regulation (EU) 2017/745 (MDR) werden folgende Anforderungen erhoben:

In diesen Formulierungen wird klar, dass Risiken während der Herstellung so weit wie möglich minimiert werden müssen. Ohne einen objektiven Nachweis, dass der Herstellprozess konstant Produkte mit Eigenschaften innerhalb der Anforderungen erfüllt, ist diese Anforderung nicht umzusetzen.In den GSPRs oder den Definitionen der GHTF Guidance / desCFR 21 Part 820.3 ist keine konkrete Angabe enthalten, wie dieser Nachweis genau auszusehen hat.In Kapitel 5 "Conduct a Validation" der GHTF Guidance sind dazu allerdings Empfehlungen/ Vorschläge enthalten.Bevor an die konkrete Umsetzung gegangen wird, muss nun folgende weitere Frage beantwortet werden:
Welche Prozesse müssen validiert werden?
Die GHTF-Guideline macht hierzu ebenfalls Vorgaben, allerdings lässt sich dazu auch etwas in der DIN EN ISO 13485:2021-12 unter 7.5.6 "Validierung der Prozesse zur Produktion und zur Dienstleistungserbringung" nachlesen. Dort ist festgelegt, dass Prozesse dann validiert werden müssen, wenn das Ergebnis nicht durch andere Maßnahmen vor Ingebrauchnahme verifiziert werden kann.Eine sehr ähnliche Anforderung lässt sich in Chapter 6 Task 7 des MDSAP Audit Approach und im CFR 21 Part 820.75 (a) finden.Ein Prozess, der ein Ergebnis beinhaltet, das zu 100 % verifiziert, also überprüft wird, z. B. ob jede Verpackung eine Gebrauchsanweisung enthält, muss also nicht validiert werden.Was ist aber mit komplexen Prozessen, bei der manche Ergebnisse gar nicht oder nur stichprobenhaft geprüft werden, die aber für andere Ergebnisse eine 100 % Kontrolle beinhalten?In solchen Fällen würde sich die Validierung auf diejenigen Eigenschaften konzentrieren, die nicht/ nicht ausreichend geprüft (verifiziert) werden. Dass diese Eigenschaften zu 100 % verifiziert werden, sollte allerdings nachvollziehbar begründet werden.
Validierung oder 100 % verifizieren?
Ob eine Validierung durchgeführt wird oder eine Kontrollmaßnahme zur 100 %-Verifizierung in den Prozess eingebaut wird, hat das Unternehmen zum Teil selbst in der Hand. Bei Eigenschaften, die zerstörend geprüft werden müssen, wie z. B. Sterilität, ist eine 100 %-Verifizierung natürlich nicht möglich. Bei anderen Eigenschaften, wie z. B. einer Füllmengenkontrolle, hat das Unternehmen aber noch die Möglichkeit diese Entscheidung zu treffen. Hier können ökonomische Beweggründe eine Rolle spielen. Dabei müssen die Kosten einer Validierung (Personalaufwand während der Validierung, Tests der Stichproben, Anlagenverfügbarkeit) den Kosten der Implementierung einer 100 % -Verifizierung gegenübergestellt werden.Die erläuterten Zusammenhänge und der Weg zur Entscheidungsfindung, ob ein Prozess validiert werden muss, sind im folgenden Schaubild abgebildet.
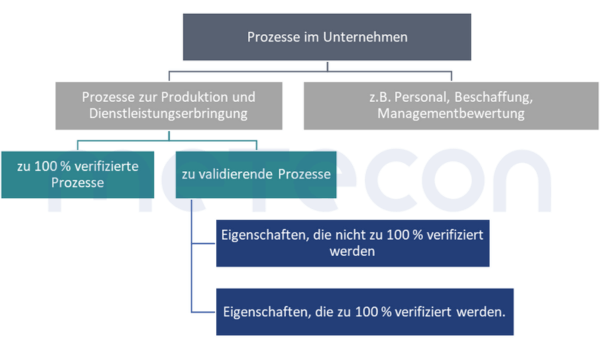
Achtung: Sofern bei Prozessen nur Teile der Eigenschaften des Produktes verändert werden, müssen auch nur die von dem Prozess betroffenen Eigenschaften bei der Validierung betrachtet werden. Dennoch kann es natürlich vorkommen, dass eine Eigenschaft unabsichtlich durch den Prozess negativ verändert wird. Solche Aspekte sollten in einer Risikoanalyse des Prozesses betrachtet und dokumentiert werden. So kann das in der Validierung genutzte Vorgehen risikobasiert gestützt begründet werden.Beispiel: Zwei Komponenten werden zusammengeschweißt. Wenn diese Schweißnaht eine sicherheitsrelevante Auswirkung auf das Produkt haben kann, muss das Ergebnis des Schweißens in der Validierung betrachtet werdenz. B. bezüglich Festigkeit. Die geometrischen Abmaße z. B. einer Bohrung, die weit von dem Schweißpunkt entfernt ist, muss nicht vermessen werden.
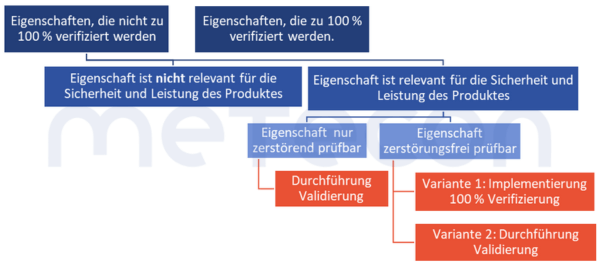
Gibt es Ausnahmen?
1. Kein Risiko für Sicherheit und LeistungEs gibt Eigenschaften, die nicht zu 100 % verifiziert werden, aber auch nicht im Rahmen einer Validierung betrachtet werden müssen. Dies ist dann der Fall, wenn von dem Fehlen oder der Abweichung von der Vorgabe kein Risiko für die Sicherheit und Leistung des Produktes ausgeht.Im MDSAP Audit Approach Chapter 6 Task 7 steht: "Processes that cannot be fully verified include processes where routine inspection and/or testing does not examine quality attributes essential to the proper functioning of the finished device…." (Zu den Prozessen, die nicht vollständig verifiziert werden können, gehören Prozesse, bei denen Routineinspektionen und/oder -tests keine Qualitätsmerkmale untersuchen, die für das ordnungsgemäße Funktionieren des fertigen Produkts wesentlich sind.)Die DIN EN ISO 13485:2021-12 fordert in 7.3.4 "Entwicklungsergebnisse", dass solche Merkmale festgelegt werden.Dies könnte zum Beispiel die Farbe eines Gehäuses sein. Gibt es solche Eigenschaften, so sollte eine solche Entscheidung allerdings begründet und dokumentiert werden.
2. Doppelaufwand nur, wo unbedingt nötigZudem existiert eine weitere Option, die den Validierungsaufwand möglicherweise reduzieren oder sogar komplett entfallen lässt. Eigenschaften, die an einer Stelle im Gesamtprozess zu100 % verifiziert werden, müssen nicht an anderer Stelle bei einer Validierung betrachtet werden. Dies greift aber nur dann, wenn die Prozesse, welche zeitlich auf die 100 % -Verifizierung folgen, mit hoher Wahrscheinlichkeit keinen negativen Einfluss auf die zu 100 % -verifizierte Eigenschaft haben. Auch solche Entscheidungen sollten nachvollziehbar dokumentiert werden, um nicht in Audits in Erklärungsnöte zu geraten.Beispiel: Ein Verpackungsprozess wird durch eine Anlage durchgeführt. Diese druckt die Etiketten und klebt diese auf die Verpackung. Es findet an der Anlage keine Kontrolle statt, ob die Daten auf den Etiketten stimmen oder ob ein Etikett auf dem Produkt vorhanden ist. Die Verpackung des etikettierten Produkts wird von Mitarbeitern zu einem späteren Zeitpunkt manuell durchgeführt. Diese müssen laut Arbeitsanweisung von jedem Produkt das Etikett einscannen und prüfen, ob die hinterlegten Daten (Charge, Produktionsdatum) korrekt sind. Ein fehlendes oder fehlerhaftes Etikett würde hierbei somit identifiziert werden.
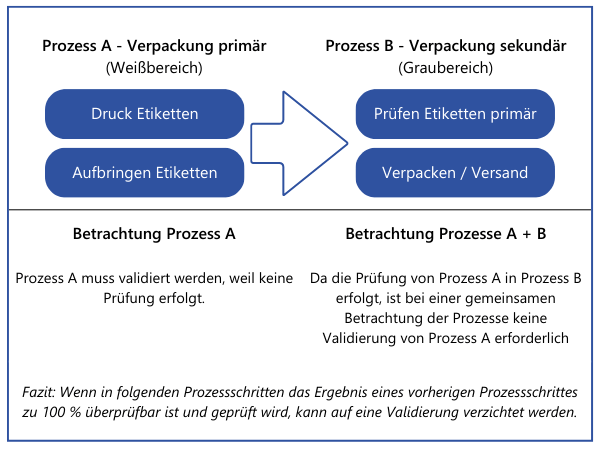
Bei dem gezeigten Vorgehen werden erst alle Prozesse zur Produktion und Dienstleistungserbringung aufgelistet und dargelegt, an welcher Stelle Eigenschaften der Produkte verändert werden, die relevant für die Sicherheit und Leistung sind. Grundsätzlich kann die Reihenfolge auch umgedreht werden. Alle Produkte und ihre sicherheits- und leistungsrelevanten Eigenschaften werden aufgelistet. Dann wird je Produkt dargelegt, welche Prozesse zur Produktion und Dienstleistungserbringung involviert sind. Letztendlich werden dieselben Informationen benötigt.Wichtig: In diesem Beitrag sprechen wir von Eigenschaften von "Produkten". Damit sind jedoch nicht nur Endprodukte gemeint. Im MDSAP Audit approachChapter 6 Task 7 wird dies explizit beschrieben: "Process validation may apply to processes that generate components, subassemblies, or finished devices". (Die Prozessvalidierung kann sich auf Prozesse beziehen, die Komponenten, Unterbaugruppen oder fertige Geräte erzeugen). Somit muss auch für diese Prozesse evaluiert werden, ob eine Validierung durchgeführt werden muss und wenn ja in welchen.
Wie entscheidet man jetzt in Summe?
In der Gesamtübersicht ergibt sich somit folgendes Entscheidungsdiagramm zur Bewertung, ob ein Prozess validiert werden muss und wenn ja, welche Eigenschaften dabei berücksichtigt werden müssen:
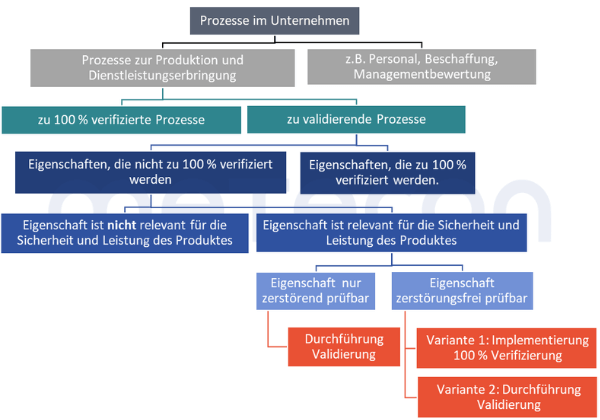
Schritt 1: Die Dokumentation der Ergebnisse
Um ein Fundament für die Validierungstätigkeiten zu schaffen, sollte ein Überblick vorhanden sein, welche Prozesse im Unternehmen durchgeführt werden und welche davon zu validieren sind. Der Begriff "Validation Master Plan" ist verbreitet für ein solches Dokument und wird von der GHTF Guidance sowie einer FDA Präsentation empfohlen. Es gibt allerdings keine regulatorische Anforderung, dass ein solches Dokument zwingend zu existieren hat. Sofern es vorhanden ist, können in ihm zusätzlich noch Verantwortlichkeiten, Zeitschienen und Verweise auf die erstellten Dokumente festgehalten werden.Und wie ging es für Frau Alid von KOMPONENTEN-KÖNIGE weiter? Vera Alid hat sich ebenfalls mit den Guidelines und Regularien vertraut gemacht und ein besseres Verständnis dafür bekommen, wie sie mit der Anfrage der Medizinprodukte GMBH umgehen soll. In unserem nächsten Beitrag erfahren Sie, wie sie unserer Meinung nach weiter vorgehen sollte.
Was Sie unbedingt mitnehmen sollten
 Leon Weißenhorn
Leon Weißenhorn
PrologDas Unternehmen KOMPONENTEN-KÖNIGE ist seit vielen Jahren erfolgreich am Markt. Es hat die Räumlichkeiten, die Anlagen und das Know-how, um am Standort Flüssigkeiten in Behälter abzufüllen. Ein Kunde ist die Medizinprodukte GmbH, für die KOMPONENTEN-KÖNIGE seit fünf Jahren einen nahezu vollautomatischen Abfüll- und Verpackungsprozess durchführt.Letzte Woche hat die Medizinprodukte GmbH telefonisch angefragt, ob Prozessvalidierungsunterlagen vorhanden seien; sie würden zeitnah benötigt! Vera Alid, Mitarbeiterin bei KOMPONENTEN-KÖNIGE, hat versprochen, sich darum zu kümmern. Nach mehreren Gesprächen mit Kolleginnen und Kollegen schwirrt Vera der Kopf: Weiß sie überhaupt, wonach genau sie sucht? Allmählich zweifelt sie daran, denn die Aussagen der Befragten sind verwirrend und manchmal widersprüchlich:
- "Eine Prozessvalidierung ist überflüssig, weil wir für jede Charge eine Prüfung am Ende machen."
- "Prozessvalidierungen müssen nur Pharma-Unternehmen machen."
- "Der Prozess läuft schon lange und ohne Probleme. Warum müssen wir jetzt Dokumente dazu vorlegen?"
- "Was sollen wir denn bei so einer Prozessvalidierung prüfen? Alles? Dafür haben wir keine Zeit!"
- "Können wir zusätzliche Kontrollen einführen, um keine Prozessvalidierung machen zu müssen?"
Validierung in der Produktion -Teil 1: Die Suche nach dem Anfang
Zunächst muss eine ganz grundlegende Frage geklärt werden:Was genau meint der Hersteller, wenn er eine Prozessvalidierung sehen will?
Gemäß der GHTF Guidance Quality Management Systems - Process Validation Guidance wird Prozessvalidierung wie folgt definiert: "establishing by objective evidence that a process consistently produces a result or product meeting its predetermined requirements." Es soll also durch objektive Nachweise festgestellt werden, dass ein Prozess kontinuierlich ein Ergebnis oder Produkt erzeugt, das seinen vorbestimmten Anforderungen entspricht. Diese Definition wurde auch von der US-amerikanischen Food and Drug Administration (FDA) in den In diesen Formulierungen wird klar, dass Risiken während der Herstellung so weit wie möglich minimiert werden müssen. Ohne einen objektiven Nachweis, dass der Herstellprozess konstant Produkte mit Eigenschaften innerhalb der Anforderungen erfüllt, ist diese Anforderung nicht umzusetzen.In den GSPRs oder den Definitionen der GHTF Guidance / des
Welche Prozesse müssen validiert werden?
Die GHTF-Guideline macht hierzu ebenfalls Vorgaben, allerdings lässt sich dazu auch etwas in der DIN EN ISO 13485:2021-12 unter 7.5.6 "Validierung der Prozesse zur Produktion und zur Dienstleistungserbringung" nachlesen. Dort ist festgelegt, dass Prozesse dann validiert werden müssen, wenn das Ergebnis nicht durch andere Maßnahmen vor Ingebrauchnahme verifiziert werden kann.Eine sehr ähnliche Anforderung lässt sich in Validierung oder 100 % verifizieren?
Ob eine Validierung durchgeführt wird oder eine Kontrollmaßnahme zur 100 %-Verifizierung in den Prozess eingebaut wird, hat das Unternehmen zum Teil selbst in der Hand. Bei Eigenschaften, die zerstörend geprüft werden müssen, wie Achtung: Sofern bei Prozessen nur Teile der Eigenschaften des Produktes verändert werden, müssen auch nur die von dem Prozess betroffenen Eigenschaften bei der Validierung betrachtet werden. Dennoch kann es natürlich vorkommen, dass eine Eigenschaft unabsichtlich durch den Prozess negativ verändert wird. Solche Aspekte sollten in einer Risikoanalyse des Prozesses betrachtet und dokumentiert werden. So kann das in der Validierung genutzte Vorgehen risikobasiert gestützt begründet werden.Beispiel: Zwei Komponenten werden zusammengeschweißt. Wenn diese Schweißnaht eine sicherheitsrelevante Auswirkung auf das Produkt haben kann, muss das Ergebnis des Schweißens in der Validierung betrachtet werden
Gibt es Ausnahmen?
1. Kein Risiko für Sicherheit und LeistungEs gibt Eigenschaften, die nicht zu 100 % verifiziert werden, aber auch nicht im Rahmen einer Validierung betrachtet werden müssen. Dies ist dann der Fall, wenn von dem Fehlen oder der Abweichung von der Vorgabe kein Risiko für die Sicherheit und Leistung des Produktes ausgeht.Im MDSAP Audit Approach 2. Doppelaufwand nur, wo unbedingt nötigZudem existiert eine weitere Option, die den Validierungsaufwand möglicherweise reduzieren oder sogar komplett entfallen lässt. Eigenschaften, die an einer Stelle im Gesamtprozess zu
Bei dem gezeigten Vorgehen werden erst alle Prozesse zur Produktion und Dienstleistungserbringung aufgelistet und dargelegt, an welcher Stelle Eigenschaften der Produkte verändert werden, die relevant für die Sicherheit und Leistung sind. Grundsätzlich kann die Reihenfolge auch umgedreht werden. Alle Produkte und ihre sicherheits- und leistungsrelevanten Eigenschaften werden aufgelistet. Dann wird je Produkt dargelegt, welche Prozesse zur Produktion und Dienstleistungserbringung involviert sind. Letztendlich werden dieselben Informationen benötigt.Wichtig: In diesem Beitrag sprechen wir von Eigenschaften von "Produkten". Damit sind jedoch nicht nur Endprodukte gemeint. Im MDSAP Audit approach
Wie entscheidet man jetzt in Summe?
In der Gesamtübersicht ergibt sich somit folgendes Entscheidungsdiagramm zur Bewertung, ob ein Prozess validiert werden muss und wenn ja, welche Eigenschaften dabei berücksichtigt werden müssen:Schritt 1: Die Dokumentation der Ergebnisse
Um ein Fundament für die Validierungstätigkeiten zu schaffen, sollte ein Überblick vorhanden sein, welche Prozesse im Unternehmen durchgeführt werden und welche davon zu validieren sind. Der Begriff "Validation Master Plan" ist verbreitet für ein solches Dokument und wird von der GHTF Guidance sowie einer FDA Präsentation empfohlen. Es gibt allerdings keine regulatorische Anforderung, dass ein solches Dokument zwingend zu existieren hat. Sofern es vorhanden ist, können in ihm zusätzlich noch Verantwortlichkeiten, Zeitschienen und Verweise auf die erstellten Dokumente festgehalten werden.Und wie ging es für Frau Alid von KOMPONENTEN-KÖNIGE weiter? Vera Alid hat sich ebenfalls mit den Guidelines und Regularien vertraut gemacht und ein besseres Verständnis dafür bekommen, wie sie mit der Anfrage der Medizinprodukte GMBH umgehen soll. In unserem nächsten Beitrag erfahren Sie, wie sie unserer Meinung nach weiter vorgehen sollte.Was Sie unbedingt mitnehmen sollten
- Die Eigenschaften, die relevant für Sicherheit und Leistung des Produktes sind, müssen bekannt sein.
- Kenntnis über die durchgeführten Prozesse und Kontrollen ist essenziell.
- Die gesamte Prozesskette für das Produkt sollte betrachtet werden. So können ggf. Eigenschaften identifiziert werden, die an anderer Stelle zu
100 % verifiziert werden. - Statt einer Validierung können noch Maßnahmen für eine
100 % Verifizierung implementiert werden. - Dokumentieren Sie ihre Entscheidungen inkl. einer Begründung.
Regulatory Affairs & Technical Documentation


 2023/607: Was steckt hinter den neuen Übergangsbestimmungen für MDR und IVDR?")